Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, Massachusetts.
Department of Medicine, Brigham and Women's Hospital, Boston, Massachusetts.
Cancer Res. 2023 Jan 18;83(2):264-284. doi: 10.1158/0008-5472.CAN-22-0423.
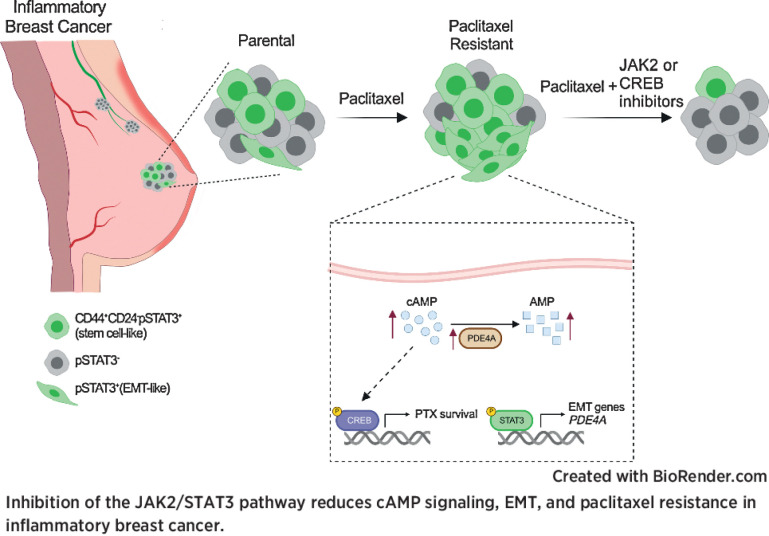
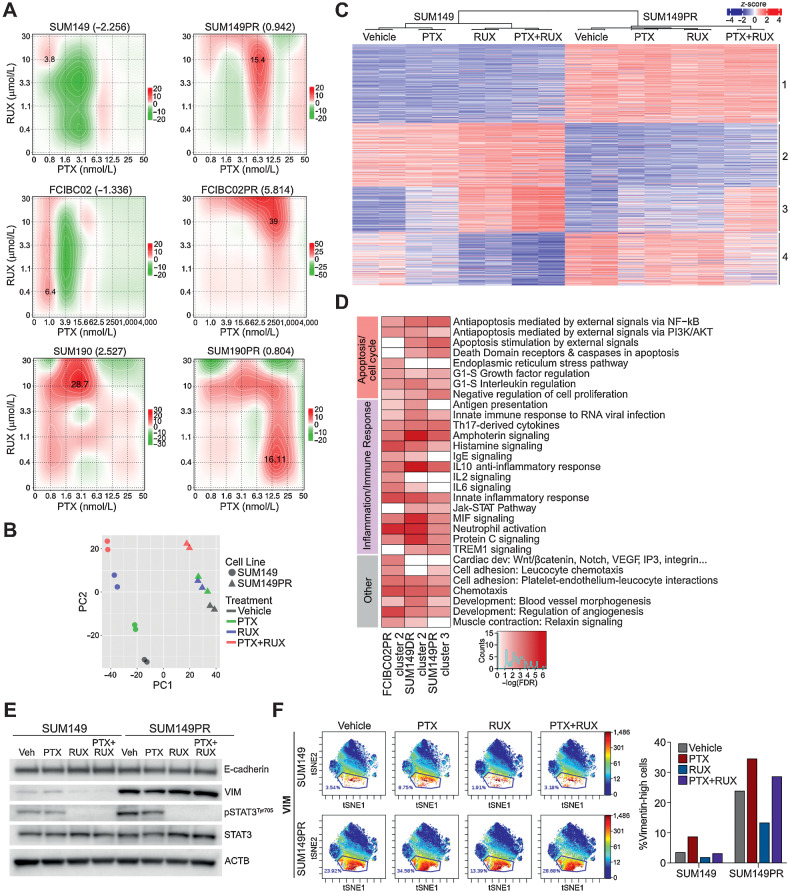
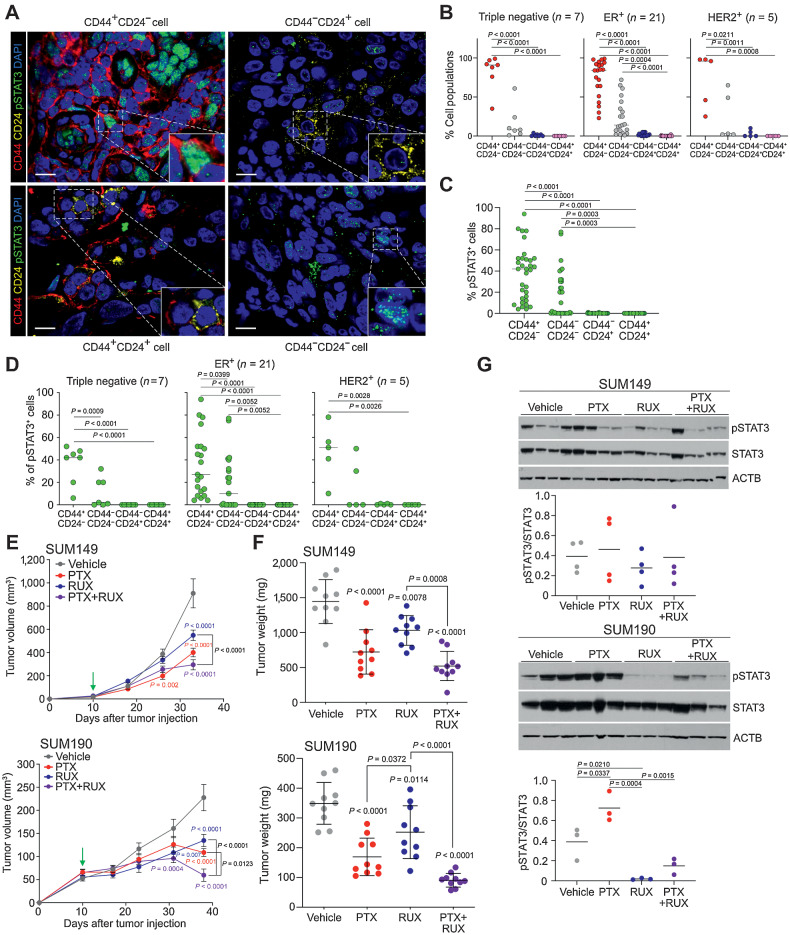
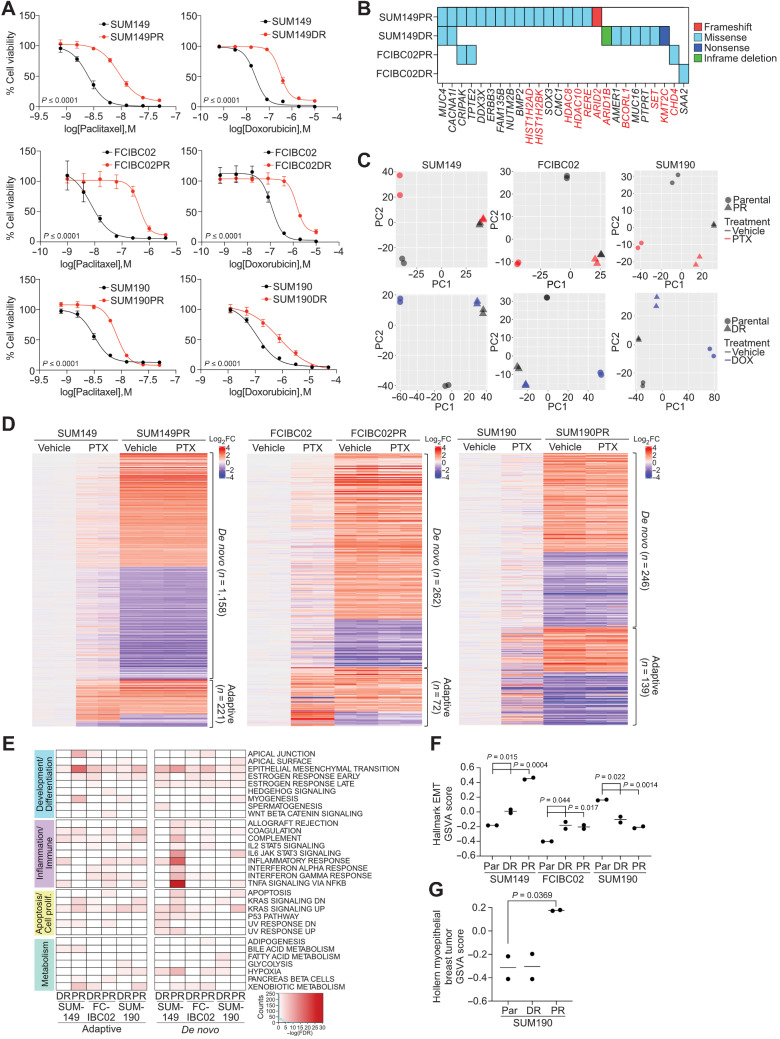
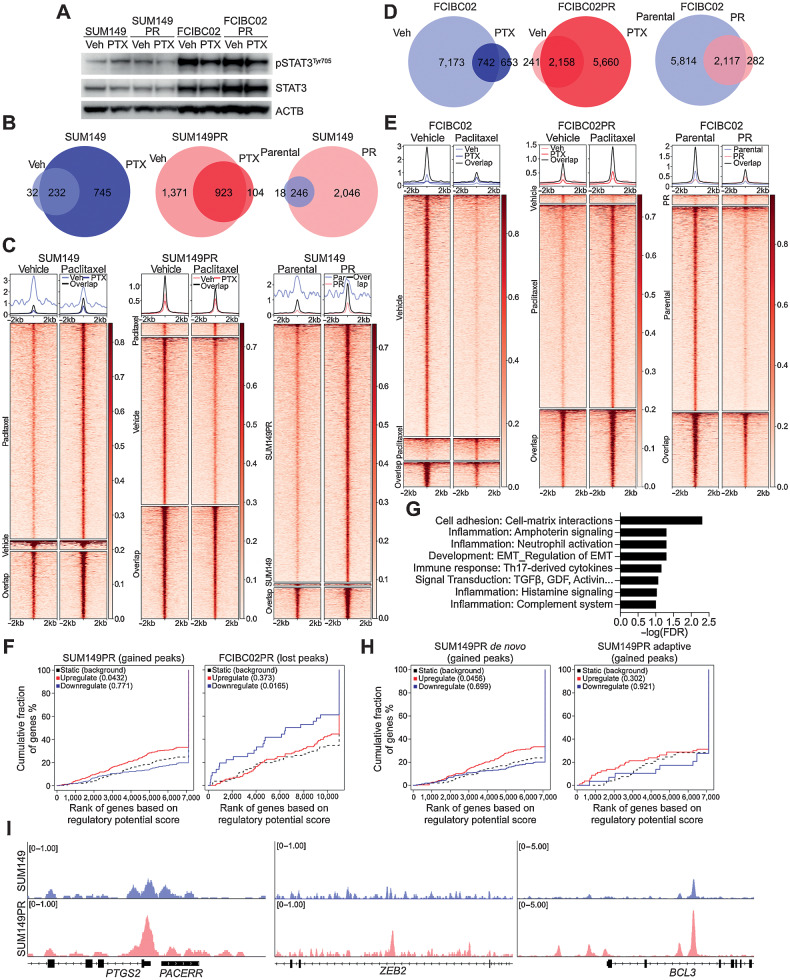
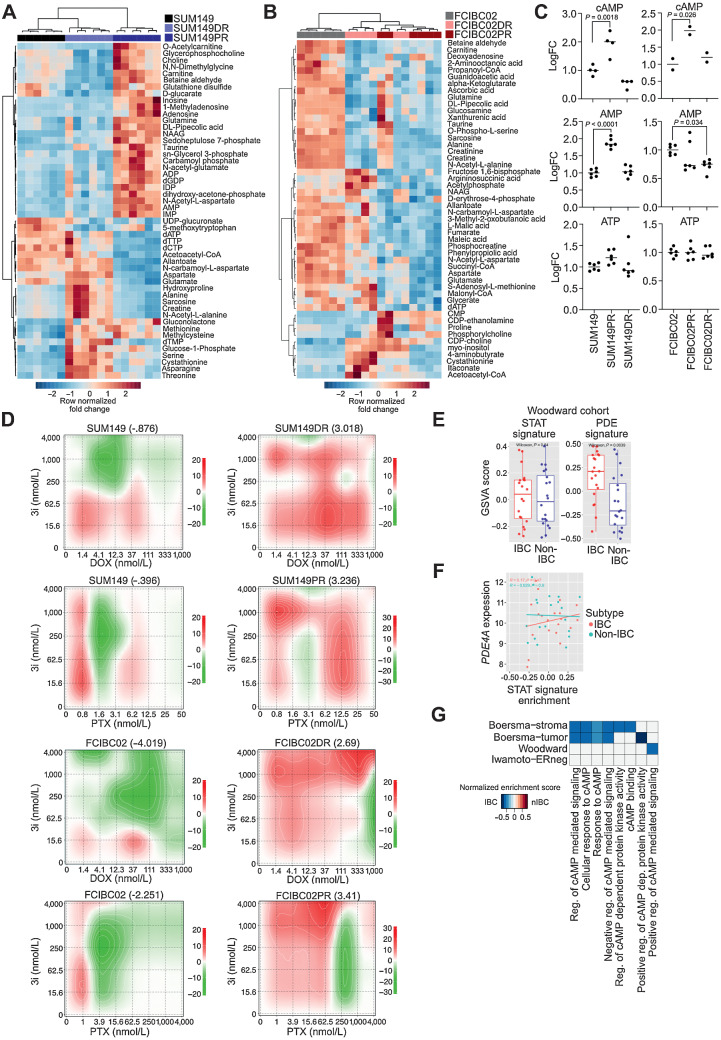
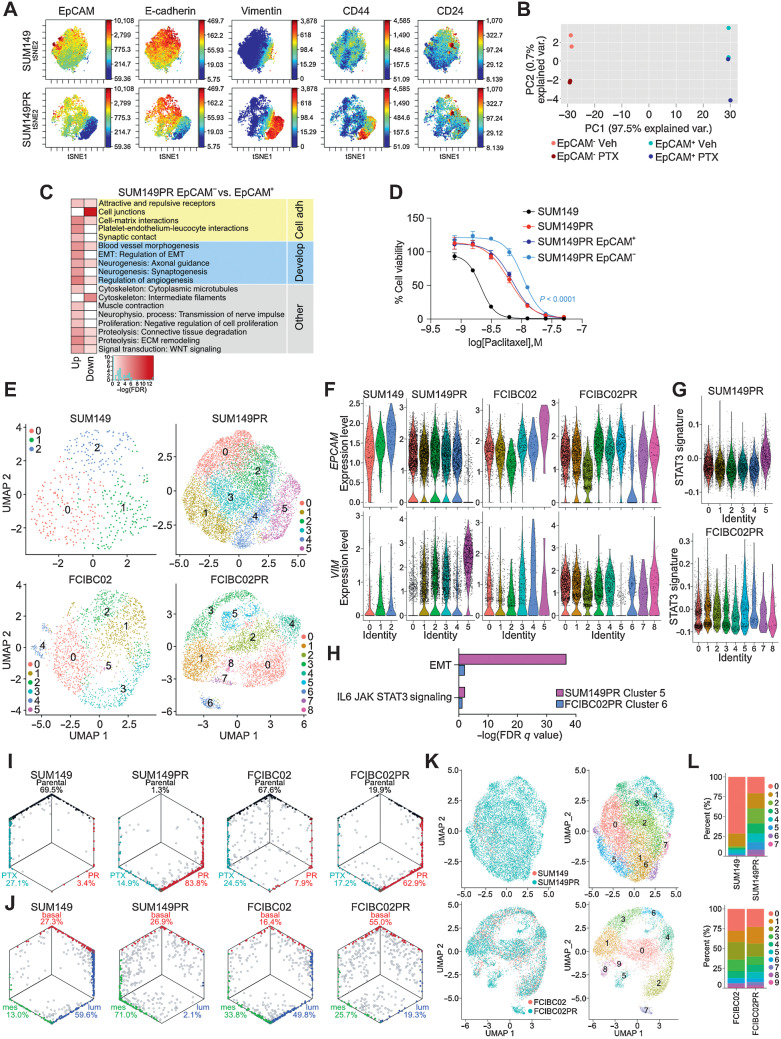
Inflammatory breast cancer (IBC) is a difficult-to-treat disease with poor clinical outcomes due to high risk of metastasis and resistance to treatment. In breast cancer, CD44+CD24- cells possess stem cell-like features and contribute to disease progression, and we previously described a CD44+CD24-pSTAT3+ breast cancer cell subpopulation that is dependent on JAK2/STAT3 signaling. Here we report that CD44+CD24- cells are the most frequent cell type in IBC and are commonly pSTAT3+. Combination of JAK2/STAT3 inhibition with paclitaxel decreased IBC xenograft growth more than either agent alone. IBC cell lines resistant to paclitaxel and doxorubicin were developed and characterized to mimic therapeutic resistance in patients. Multi-omic profiling of parental and resistant cells revealed enrichment of genes associated with lineage identity and inflammation in chemotherapy-resistant derivatives. Integrated pSTAT3 chromatin immunoprecipitation sequencing and RNA sequencing (RNA-seq) analyses showed pSTAT3 regulates genes related to inflammation and epithelial-to-mesenchymal transition (EMT) in resistant cells, as well as PDE4A, a cAMP-specific phosphodiesterase. Metabolomic characterization identified elevated cAMP signaling and CREB as a candidate therapeutic target in IBC. Investigation of cellular dynamics and heterogeneity at the single cell level during chemotherapy and acquired resistance by CyTOF and single cell RNA-seq identified mechanisms of resistance including a shift from luminal to basal/mesenchymal cell states through selection for rare preexisting subpopulations or an acquired change. Finally, combination treatment with paclitaxel and JAK2/STAT3 inhibition prevented the emergence of the mesenchymal chemo-resistant subpopulation. These results provide mechanistic rational for combination of chemotherapy with inhibition of JAK2/STAT3 signaling as a more effective therapeutic strategy in IBC.
Chemotherapy resistance in inflammatory breast cancer is driven by the JAK2/STAT3 pathway, in part via cAMP/PKA signaling and a cell state switch, which can be overcome using paclitaxel combined with JAK2 inhibitors.
炎性乳腺癌(IBC)是一种难以治疗的疾病,由于转移风险高和治疗耐药性差,临床结局较差。在乳腺癌中,CD44+CD24-细胞具有干细胞样特征,并促进疾病进展,我们之前描述了一种依赖于 JAK2/STAT3 信号的 CD44+CD24-pSTAT3+乳腺癌细胞亚群。在这里,我们报告 CD44+CD24-细胞是 IBC 中最常见的细胞类型,并且通常是 pSTAT3+。JAK2/STAT3 抑制与紫杉醇联合使用可使 IBC 异种移植物生长减少超过任一药物单独使用。开发并表征对紫杉醇和多柔比星耐药的 IBC 细胞系,以模拟患者的治疗耐药性。对亲本和耐药细胞进行多组学分析显示,在化疗耐药衍生物中富集与谱系身份和炎症相关的基因。整合 pSTAT3 染色质免疫沉淀测序和 RNA 测序(RNA-seq)分析表明,pSTAT3 调节耐药细胞中与炎症和上皮-间充质转化(EMT)相关的基因,以及 PDE4A,一种 cAMP 特异性磷酸二酯酶。代谢组学特征鉴定出在 IBC 中升高的 cAMP 信号和 CREB 作为候选治疗靶点。通过 CyTOF 和单细胞 RNA-seq 在化疗和获得性耐药过程中对细胞动力学和异质性进行单细胞水平的研究,确定了耐药机制,包括通过选择罕见的预先存在的亚群或获得性变化,从腔细胞状态向基底/间充质细胞状态的转变。最后,紫杉醇和 JAK2/STAT3 抑制联合治疗可防止出现间充质化疗耐药亚群。这些结果为化疗联合 JAK2/STAT3 信号抑制作为 IBC 更有效的治疗策略提供了机制合理性。
炎性乳腺癌的化疗耐药性是由 JAK2/STAT3 通路驱动的,部分通过 cAMP/PKA 信号和细胞状态转换,使用紫杉醇联合 JAK2 抑制剂可以克服这种耐药性。