Department of Medicine, Queen's University, Kingston, ON, Canada.
Division of Genetic Medicine, Vanderbilt University Medical Center, Nashville, TN.
Blood. 2023 May 4;141(18):2214-2223. doi: 10.1182/blood.2022018825.
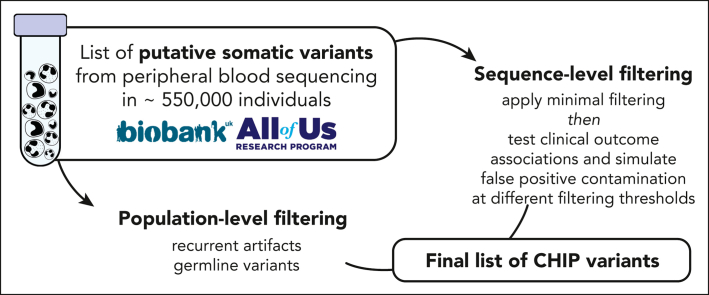
Clonal hematopoiesis of indeterminate potential (CHIP) is a common form of age-related somatic mosaicism that is associated with significant morbidity and mortality. CHIP mutations can be identified in peripheral blood samples that are sequenced using approaches that cover the whole genome, the whole exome, or targeted genetic regions; however, differentiating true CHIP mutations from sequencing artifacts and germ line variants is a considerable bioinformatic challenge. We present a stepwise method that combines filtering based on sequencing metrics, variant annotation, and population-based associations to increase the accuracy of CHIP calls. We apply this approach to ascertain CHIP in ∼550 000 individuals in the UK Biobank complete whole exome cohort and the All of Us Research Program initial whole genome release cohort. CHIP ascertainment on this scale unmasks recurrent artifactual variants and highlights the importance of specialized filtering approaches for several genes, including TET2 and ASXL1. We show how small changes in filtering parameters can considerably increase CHIP misclassification and reduce the effect size of epidemiological associations. Our high-fidelity call set refines previous population-based associations of CHIP with incident outcomes. For example, the annualized incidence of myeloid malignancy in individuals with small CHIP clones is 0.03% per year, which increases to 0.5% per year among individuals with very large CHIP clones. We also find a significantly lower prevalence of CHIP in individuals of self-reported Latino or Hispanic ethnicity in All of Us, highlighting the importance of including diverse populations. The standardization of CHIP calling will increase the fidelity of CHIP epidemiological work and is required for clinical CHIP diagnostic assays.
不确定潜能的克隆性造血 (CHIP) 是一种常见的与重大发病率和死亡率相关的与年龄相关的体细胞镶嵌现象。CHIP 突变可以在使用覆盖全基因组、全外显子或靶向遗传区域的方法对测序的外周血样本中识别出来;然而,将真正的 CHIP 突变与测序伪影和种系变异区分开来是一个相当大的生物信息学挑战。我们提出了一种逐步的方法,该方法结合了基于测序指标、变体注释和基于群体的关联的过滤,以提高 CHIP 调用的准确性。我们将这种方法应用于 UK Biobank 完整外显子组队列和 All of Us Research Program 初始全基因组发布队列中约 550000 个人的 CHIP 确定。这种规模的 CHIP 确定揭示了反复出现的人为变体,并强调了针对包括 TET2 和 ASXL1 在内的几个基因的专门过滤方法的重要性。我们展示了过滤参数的微小变化如何极大地增加 CHIP 分类错误并降低流行病学关联的效应大小。我们的高保真呼叫集细化了之前基于人群的 CHIP 与发病结果的关联。例如,个体中具有小 CHIP 克隆的髓样恶性肿瘤的年化发病率为每年 0.03%,而在具有非常大 CHIP 克隆的个体中,发病率增加到每年 0.5%。我们还发现,在 All of Us 中,自我报告的拉丁裔或西班牙裔个体中 CHIP 的患病率明显较低,这突出了纳入不同人群的重要性。CHIP 调用的标准化将提高 CHIP 流行病学工作的保真度,并且是临床 CHIP 诊断检测所必需的。