Kuninobu Ken-Ichiro, Takemura Taichiro, Takizawa Yu, Hasebe Futoshi, Yamashiro Tetsu
Graduate School of Biomedical Sciences, Nagasaki University, 1-12-4 Sakamoto Nagasaki city, Nagasaki, 852-8523, Japan.
Vietnam Research Station, Institute of Tropical Medicine, Nagasaki University, 1-12-4 Sakamoto Nagasaki city, Nagasaki, 852-8523, Japan.
Trop Med Health. 2023 Feb 2;51(1):5. doi: 10.1186/s41182-023-00500-4.
Cholera is a water-borne disease caused by toxigenic Vibrio cholerae serogroups O1 and O139. Not a few studies on the whole-genome analyses of V. cholerae O1 biotype El Tor have been published; however, the number of analyses for biotype classical is limited. The whole-genome analysis was made on a V. cholerae biotype classical strain, Man9, isolated in 1946 in Sasebo city, Nagasaki prefecture, from a returnee from the northeast part of China.
PacBio RSII was used to determine the whole-genome of Man9. De novo assemblies were made with CLC Genomics Workbench 8.5.1 and Canu. 2.0 and annotated by Prokka version 1.12. Upon determining the configuration of the CTX prophage region, combined procedures of PCR, RFLP with Southern blotting, and Sanger sequencing method were used. The phylogenetic tree was constructed by RaxML and visualized by Phandango. The identification of Cas genes and spacer sequences was made by CRISPR-finder and NCBI Blast search. These data were compared with those of V. cholerae serogroup O1 biotype classical O395.
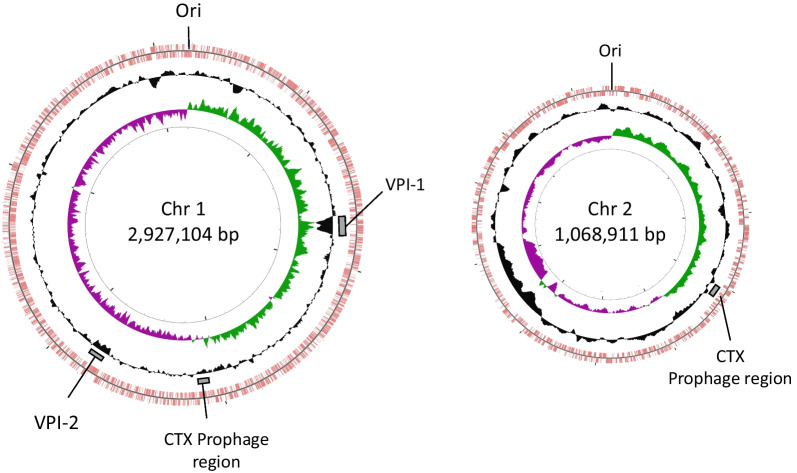
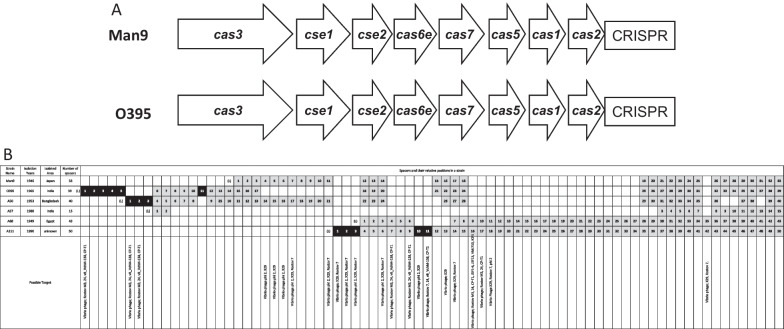
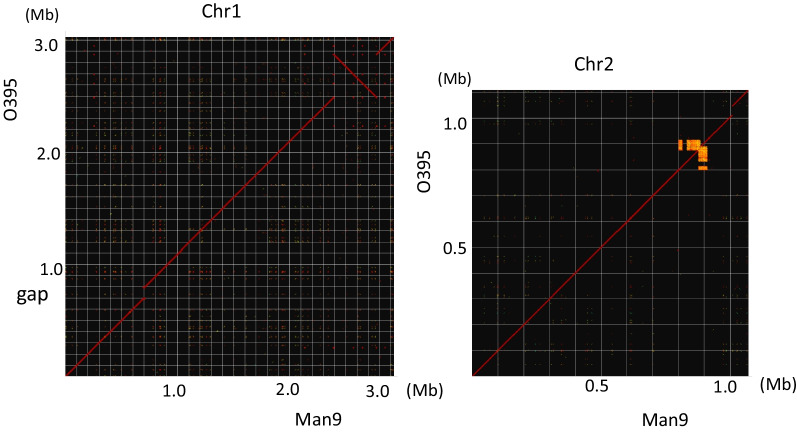
The Man9 carried the 2.9 Mb (Chr1) and 1.1 Mb (Chr2) chromosomes with 2683 and 1198 CDSs, respectively. The genome similarity between Man9 and O395 was 97.0% when the total genomes were compared. Man9 carried a 380-kb inversion on the Chr1, and 95-kb and 35-kb fragments were not present on the Chr1 and on the Chr2, respectively. Man9 monophyletically clustered with 23 other biotype classical strains on the core gene phylogenetic tree analyses. Man9 carries "CTX" and a stretch of "truncated CTX-CTX" on the Chr1 and the Chr2, respectively, which is the opposite arrangement of O395. Man9 carries CRISPR-Cas system subtype I-E with 33 spacers, 64% of which were identical to those of O395.
Man9 differs from O395 by 3% on the total genome comparison; however, genomic analysis of a strain having circulated in the interpandemic period between the 6th and the 7th cholera pandemic is valuable and contributes to understanding the evolution of pathogenic V. cholerae.
霍乱是一种由产毒性霍乱弧菌O1群和O139群引起的水传播疾病。关于霍乱弧菌O1生物型埃尔托的全基因组分析已有不少研究发表;然而,对古典生物型的分析数量有限。对1946年从中国东北地区归国人员中分离于长崎县佐世保市的一株霍乱弧菌古典生物型菌株Man9进行了全基因组分析。
使用PacBio RSII测定Man9的全基因组。使用CLC Genomics Workbench 8.5.1和Canu 2.0进行从头组装,并用Prokka 1.12版本进行注释。在确定CTX噬菌体区域的结构后,采用PCR、Southern杂交RFLP和桑格测序法相结合的方法。通过RaxML构建系统发育树,并通过Phandango进行可视化。通过CRISPR-finder和NCBI Blast搜索鉴定Cas基因和间隔序列。将这些数据与霍乱弧菌O1血清群古典生物型O395的数据进行比较。
Man9携带2.9 Mb(Chr1)和1.1 Mb(Chr2)染色体,分别有2683个和1198个编码序列。比较全基因组时,Man9与O395之间的基因组相似性为97.0%。Man9在Chr1上有一个380 kb的倒位,Chr1和Chr2上分别不存在95 kb和35 kb的片段。在核心基因系统发育树分析中,Man9与其他23株古典生物型菌株单系聚类。Man9在Chr1和Chr2上分别携带“CTX”和一段“截短的CTX-CTX”,这与O395的排列相反。Man9携带CRISPR-Cas系统I-E亚型,有33个间隔序列,其中64%与O395的相同。
在全基因组比较中,Man9与O395有3%的差异;然而,对一株在第六次和第七次霍乱大流行期间的流行间期传播的菌株进行基因组分析是有价值的,有助于了解致病性霍乱弧菌的进化。