Du Xin-Jie, Yang Xian-Rong, Wang Qi-Cai, Lin Guo-Liang, Li Peng-Fei, Zhang Wei-Feng
Department of Thyroid and Breast Surgery, LongYan First Hospital, Longyan, 364000, Fujian, China.
Department of General Surgery, Linhai Hospital of Traditional Chinese Medicine, Linhai, 317000, Zhejiang, China.
Heliyon. 2023 Jan 27;9(2):e13185. doi: 10.1016/j.heliyon.2023.e13185. eCollection 2023 Feb.
This study aimed to identify prognostic signatures to predict the prognosis of breast cancer (BRCA) patients based on a series of comprehensive analyses of gene expression data.
The RNA-sequencing expression data and corresponding BRCA patient clinical data were collected from the Cancer Genome Atlas (TCGA) and the Gene Expression Omnibus (GEO) datasets. Firstly, the differently expressed genes (DEGs) related to prognosis between tumor tissues and normal tissues were ascertained by performing R package "limma". Secondly, the DEGs were used to construct a polygenic risk scoring model by the weighted gene co-expression network analysis (WGCNA) and the least absolute shrinkage and selection operator Cox regression (Lasso-cox) analysis method. Thirdly, survival analysis was performed to investigate the risk score values in the TCGA cohort. And the enrichment analysis, immune cell infiltration levels analysis, and protein-protein internet (PPI) analysis were performed. Simultaneously, the GEO cohort was used to validate the model. Lastly, we constructed a nomogram to explore the influence of polygenic risk score and other clinical factors on the survival probability of patients with BRCA.
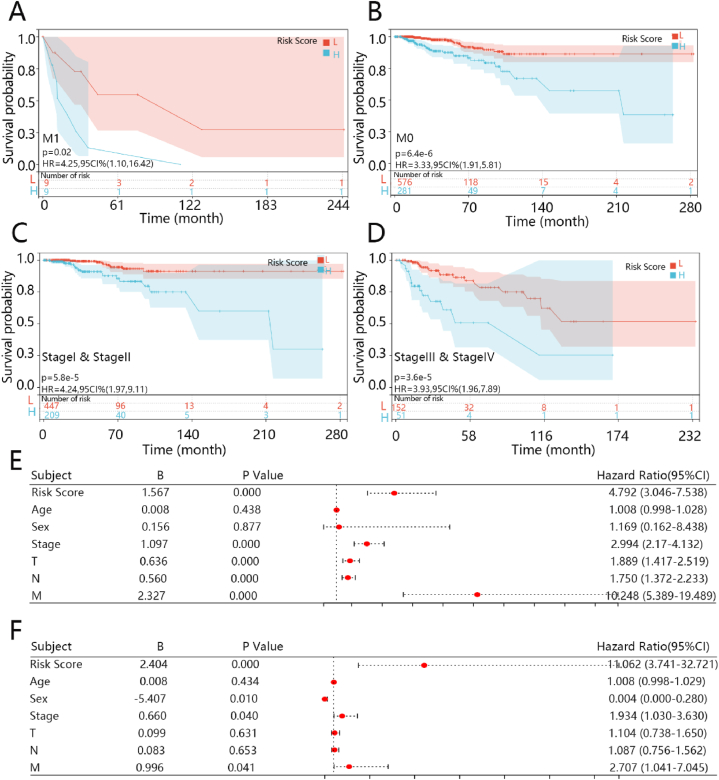
A total of 1000 DEGs including 396 upregulated genes and 604 downregulated genes were identified from the TCGA-BRCA dataset. We obtained 5 prognosis-related genes, as the key biomarkers by Lasso-cox analysis (, , , , and ), all of which were significantly upregulated in breast tumors. The prognostic prediction of the 5 genes model was great in training and validation cohorts. Moreover, the high-risk group had a poorer prognosis. The Cox regression analysis showed that the comprehensive risk score for 5 genes was an independent prognosis factor.
The 5 genes risk model constructed in this study had an independent predictive ability to distinguish patients with a high risk of death from those with a low-risk score, and it can be used as a practical and reliable prognostic tool for BRCA.
本研究旨在通过对基因表达数据进行一系列综合分析,确定预测乳腺癌(BRCA)患者预后的预后特征。
从癌症基因组图谱(TCGA)和基因表达综合数据库(GEO)数据集中收集RNA测序表达数据及相应的BRCA患者临床数据。首先,通过运行R包“limma”确定肿瘤组织与正常组织之间与预后相关的差异表达基因(DEG)。其次,利用加权基因共表达网络分析(WGCNA)和最小绝对收缩和选择算子Cox回归(Lasso-cox)分析方法,将这些DEG用于构建多基因风险评分模型。第三,进行生存分析以研究TCGA队列中的风险评分值。并进行富集分析、免疫细胞浸润水平分析和蛋白质-蛋白质相互作用(PPI)分析。同时,使用GEO队列验证该模型。最后,我们构建了列线图以探讨多基因风险评分和其他临床因素对BRCA患者生存概率的影响。
从TCGA-BRCA数据集中共鉴定出1000个DEG,其中包括396个上调基因和604个下调基因。通过Lasso-cox分析,我们获得了5个与预后相关的基因作为关键生物标志物(、、、和),所有这些基因在乳腺肿瘤中均显著上调。5基因模型在训练和验证队列中的预后预测效果良好。此外,高风险组的预后较差。Cox回归分析表明,5个基因的综合风险评分是一个独立的预后因素。
本研究构建的5基因风险模型具有独立的预测能力,可区分高死亡风险患者和低风险评分患者,可作为BRCA实用且可靠的预后工具。