Seshadri Rekha, Roux Simon, Huber Katharina J, Wu Dongying, Yu Sora, Udwary Dan, Call Lee, Nayfach Stephen, Hahnke Richard L, Pukall Rüdiger, White James R, Varghese Neha J, Webb Cody, Palaniappan Krishnaveni, Reimer Lorenz C, Sardà Joaquim, Bertsch Jonathon, Mukherjee Supratim, Reddy T B K, Hajek Patrick P, Huntemann Marcel, Chen I-Min A, Spunde Alex, Clum Alicia, Shapiro Nicole, Wu Zong-Yen, Zhao Zhiying, Zhou Yuguang, Evtushenko Lyudmila, Thijs Sofie, Stevens Vincent, Eloe-Fadrosh Emiley A, Mouncey Nigel J, Yoshikuni Yasuo, Whitman William B, Klenk Hans-Peter, Woyke Tanja, Göker Markus, Kyrpides Nikos C, Ivanova Natalia N
US Department of Energy Joint Genome Institute, Berkeley, CA, USA.
Leibniz Institute DSMZ - German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany.
Cell Genom. 2022 Nov 11;2(12):100213. doi: 10.1016/j.xgen.2022.100213. eCollection 2022 Dec 14.
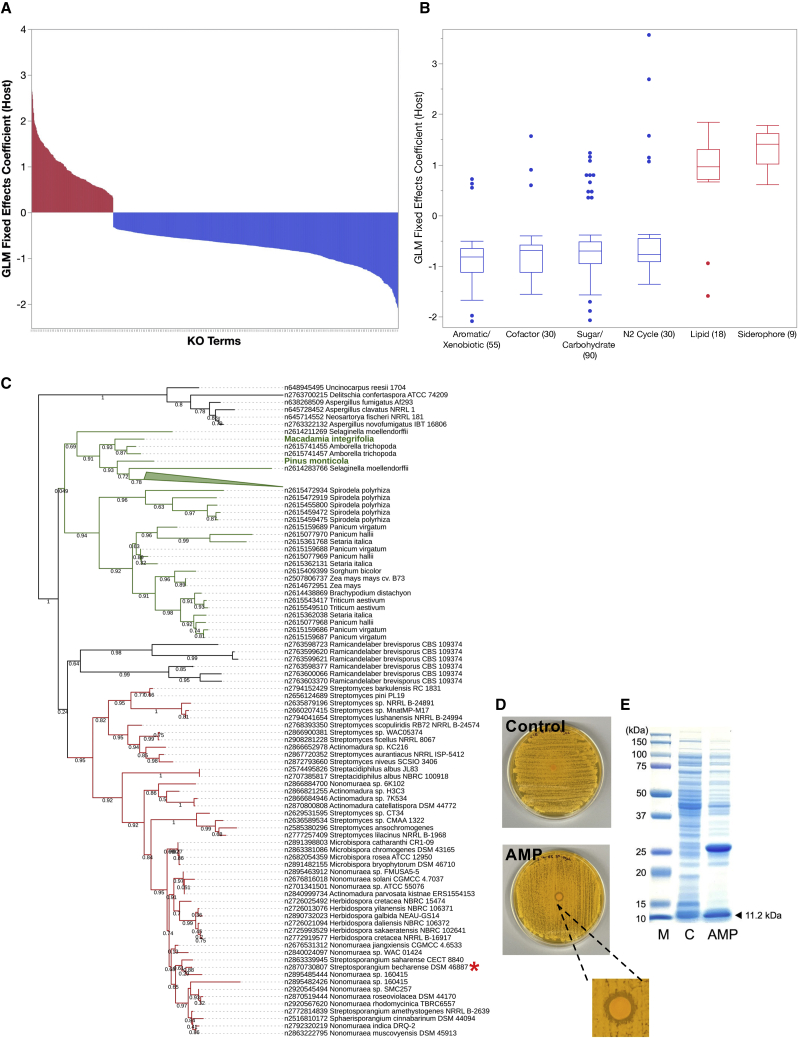
The phylum includes important human pathogens like and and renowned producers of secondary metabolites of commercial interest, yet only a small part of its diversity is represented by sequenced genomes. Here, we present 824 actinobacterial isolate genomes in the context of a phylum-wide analysis of 6,700 genomes including public isolates and metagenome-assembled genomes (MAGs). We estimate that only 30%-50% of projected actinobacterial phylogenetic diversity possesses genomic representation via isolates and MAGs. A comparison of gene functions reveals novel determinants of host-microbe interaction as well as environment-specific adaptations such as potential antimicrobial peptides. We identify plasmids and prophages across isolates and uncover extensive prophage diversity structured mainly by host taxonomy. Analysis of >80,000 biosynthetic gene clusters reveals that horizontal gene transfer and gene loss shape secondary metabolite repertoire across taxa. Our observations illustrate the essential role of and need for high-quality isolate genome sequences.
该门包括重要的人类病原体,如[具体病原体1]和[具体病原体2],以及具有商业价值的次生代谢产物的著名生产者,但只有一小部分的多样性通过测序基因组得到体现。在此,我们在对6700个基因组(包括公共分离株和宏基因组组装基因组(MAG))进行全门分析的背景下,展示了824个放线菌分离株基因组。我们估计,预计的放线菌系统发育多样性中只有30%-50%通过分离株和MAG获得了基因组代表性。基因功能比较揭示了宿主-微生物相互作用的新决定因素以及特定环境适应性,如潜在的抗菌肽。我们在分离株中鉴定出质粒和原噬菌体,并发现主要由宿主分类学构建的广泛的原噬菌体多样性。对80000多个生物合成基因簇的分析表明,水平基因转移和基因丢失塑造了不同分类群的次生代谢产物库。我们的观察结果说明了高质量分离株基因组序列的重要作用和必要性。