Fitzgerald Tomas, Birney Ewan
European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), Wellcome Genome Campus, Cambridge CB10 1SD, UK.
Cell Genom. 2022 Aug 10;2(8):100167. doi: 10.1016/j.xgen.2022.100167.
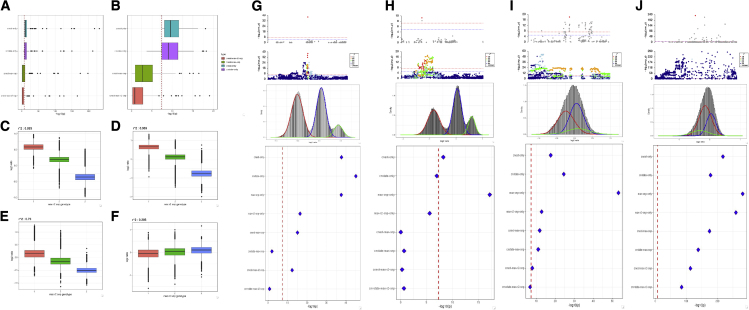
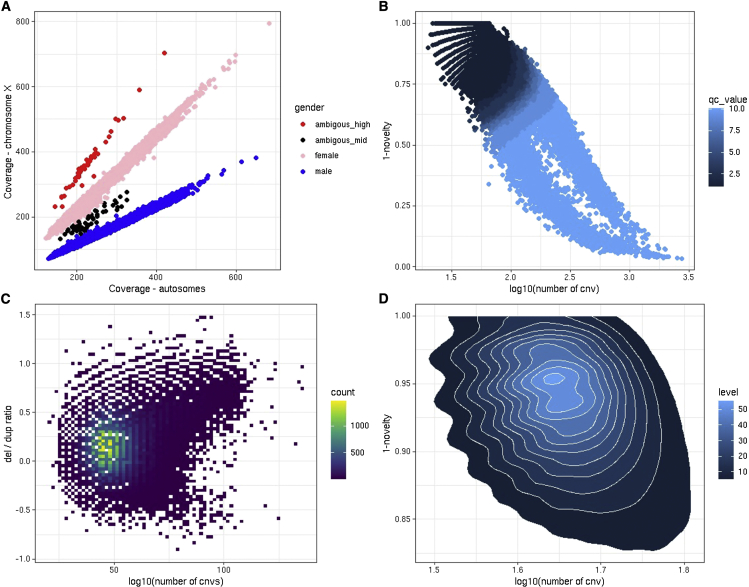
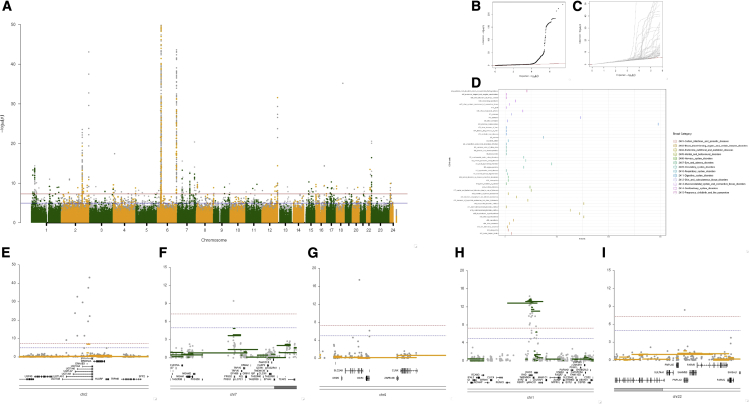
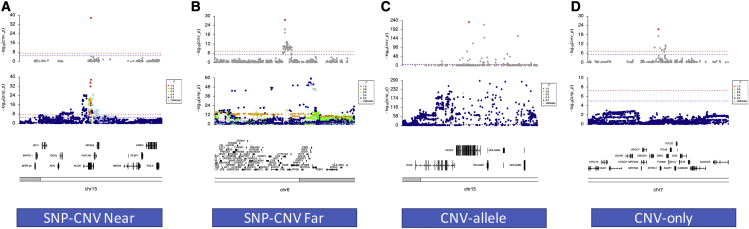
Copy number variation (CNV) is known to influence human traits, having a rich history of research into common and rare genetic disease, and although CNV is accepted as an important class of genomic variation, progress on copy-number-based genome-wide association studies (GWASs) from next-generation sequencing (NGS) data has been limited. Here we present a novel method for large-scale copy number analysis from NGS data generating robust copy number estimates and allowing copy number GWASs (CN-GWASs) to be performed genome-wide in discovery mode. We provide a detailed analysis in the UK Biobank resource and a specifically designed software package. We use these methods to perform CN-GWAS analysis across 78 human traits, discovering over 800 genetic associations that are likely to contribute strongly to trait distributions. Finally, we compare CNV and SNP association signals across the same traits and samples, defining specific CNV association classes.
已知拷贝数变异(CNV)会影响人类性状,在常见和罕见遗传病研究方面有着丰富的历史,尽管CNV被公认为一类重要的基因组变异,但基于下一代测序(NGS)数据的基于拷贝数的全基因组关联研究(GWAS)进展有限。在此,我们提出了一种从NGS数据进行大规模拷贝数分析的新方法,该方法能生成可靠的拷贝数估计值,并允许在发现模式下在全基因组范围内进行拷贝数GWAS(CN-GWAS)。我们在英国生物银行资源中进行了详细分析,并提供了一个专门设计的软件包。我们使用这些方法对78种人类性状进行CN-GWAS分析,发现了800多个可能对性状分布有重大贡献的遗传关联。最后,我们比较了相同性状和样本中的CNV和SNP关联信号,定义了特定的CNV关联类别。