Zheng Wei, Tsompana Maria, Ruscitto Angela, Sharma Ashu, Genco Robert, Sun Yijun, Buck Michael J
Department of Computer Science and Engineering, State University of New York at Buffalo, Buffalo, NY, 14203, USA.
Center of Excellence in Bioinformatics and Life Sciences, State University of New York at Buffalo, Buffalo, NY, 14203, USA.
Microbiome. 2015 Oct 5;3:48. doi: 10.1186/s40168-015-0110-9.
Currently, taxonomic interrogation of microbiota is based on amplification of 16S rRNA gene sequences in clinical and scientific settings. Accurate evaluation of the microbiota depends heavily on the primers used, and genus/species resolution bias can arise with amplification of non-representative genomic regions. The latest Illumina MiSeq sequencing chemistry has extended the read length to 300 bp, enabling deep profiling of large number of samples in a single paired-end reaction at a fraction of the cost. An increasingly large number of researchers have adopted this technology for various microbiome studies targeting the 16S rRNA V3-V4 hypervariable region.
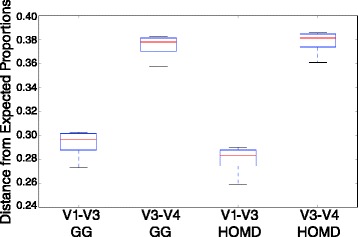
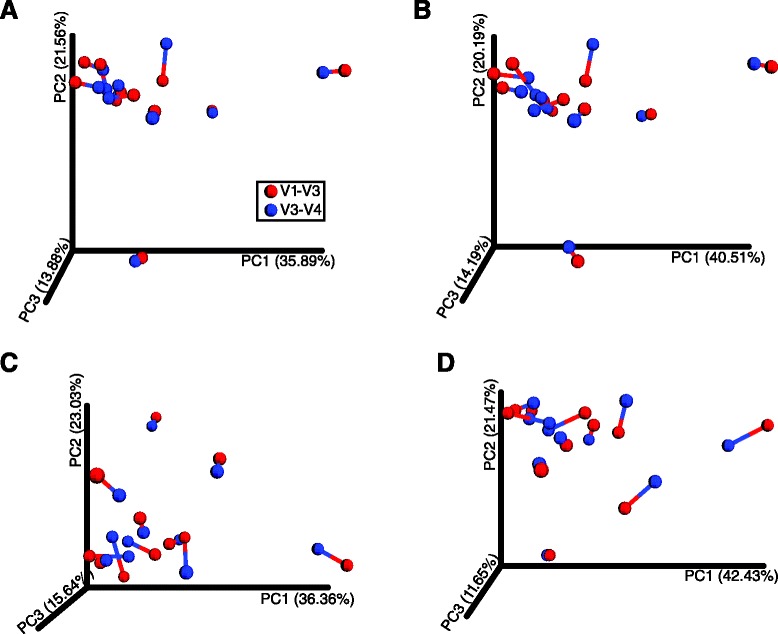
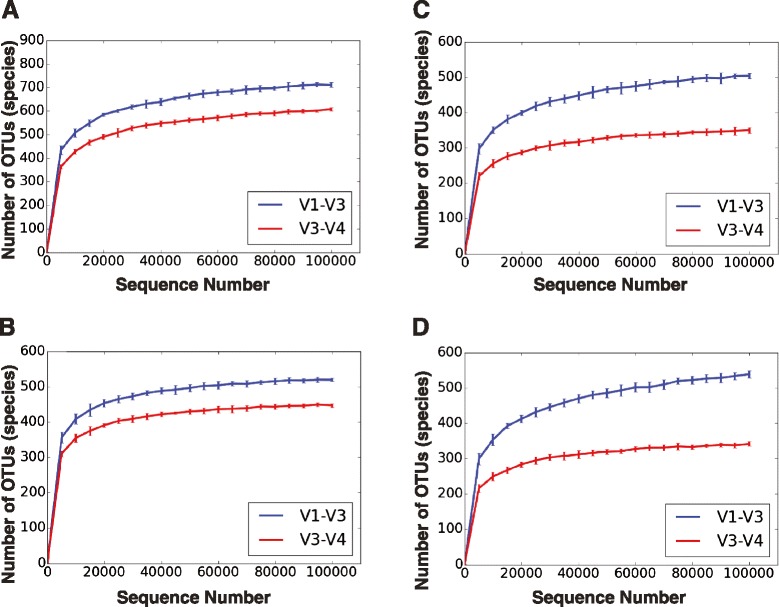
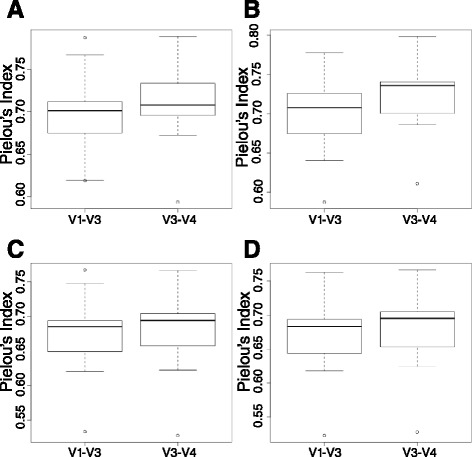
To expand the applicability of this powerful platform for further descriptive and functional microbiome studies, we standardized and tested an efficient, reliable, and straightforward workflow for the amplification, library construction, and sequencing of the 16S V1-V3 hypervariable region using the new 2 × 300 MiSeq platform. Our analysis involved 11 subgingival plaque samples from diabetic and non-diabetic human subjects suffering from periodontitis. The efficiency and reliability of our experimental protocol was compared to 16S V3-V4 sequencing data from the same samples. Comparisons were based on measures of observed taxonomic richness and species evenness, along with Procrustes analyses using beta(β)-diversity distance metrics. As an experimental control, we also analyzed a total of eight technical replicates for the V1-V3 and V3-V4 regions from a synthetic community with known bacterial species operon counts. We show that our experimental protocol accurately measures true bacterial community composition. Procrustes analyses based on unweighted UniFrac β-diversity metrics depicted significant correlation between oral bacterial composition for the V1-V3 and V3-V4 regions. However, measures of phylotype richness were higher for the V1-V3 region, suggesting that V1-V3 offers a deeper assessment of population diversity and community ecology for the complex oral microbiota.
This study provides researchers with valuable experimental evidence for the selection of appropriate 16S amplicons for future human oral microbiome studies. We expect that the tested 16S V1-V3 framework will be widely applicable to other types of microbiota, allowing robust, time-efficient, and inexpensive examination of thousands of samples for population, phylogenetic, and functional crossectional and longitutidal studies.
目前,在临床和科研环境中,对微生物群进行分类学分析是基于16S rRNA基因序列的扩增。微生物群的准确评估在很大程度上取决于所使用的引物,并且非代表性基因组区域的扩增可能会导致属/种分辨率偏差。最新的Illumina MiSeq测序技术将读长延长至了300bp,使得在单次双端反应中以较低成本对大量样本进行深度分析成为可能。越来越多的研究人员已将该技术应用于针对16S rRNA V3-V4高变区的各种微生物组研究。
为了扩展这个强大平台在进一步描述性和功能性微生物组研究中的适用性,我们使用新的2×300 MiSeq平台,对16S V1-V3高变区的扩增、文库构建和测序流程进行了标准化,并测试了一种高效、可靠且简单的工作流程。我们的分析涉及11份来自患有牙周炎的糖尿病和非糖尿病人类受试者的龈下菌斑样本。我们将实验方案的效率和可靠性与相同样本的16S V3-V4测序数据进行了比较。比较基于观察到的分类学丰富度和物种均匀度的测量,以及使用β多样性距离度量的普氏分析。作为实验对照,我们还分析了来自具有已知细菌种类操纵子计数的合成群落的V1-V3和V3-V4区域的总共8个技术重复样本。我们表明,我们的实验方案能够准确测量真实的细菌群落组成。基于未加权UniFrac β多样性度量的普氏分析表明,V1-V3和V3-V4区域的口腔细菌组成之间存在显著相关性。然而,V1-V3区域的系统发育型丰富度测量值更高,这表明V1-V3为复杂的口腔微生物群的种群多样性和群落生态学提供了更深入的评估。
本研究为研究人员在未来人类口腔微生物组研究中选择合适的16S扩增子提供了有价值的实验证据。我们预计,经过测试的16S V1-V3框架将广泛适用于其他类型的微生物群,从而能够对数千个样本进行强大、高效且经济的检查,以用于种群、系统发育以及功能的横断面和纵向研究。