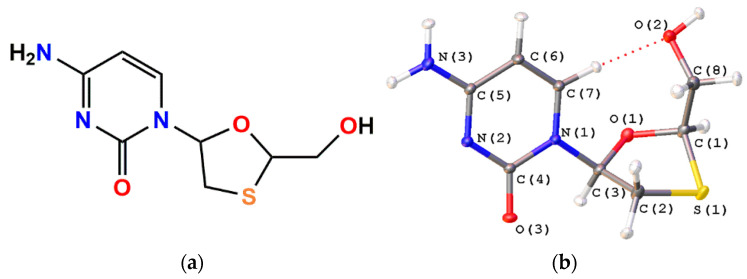
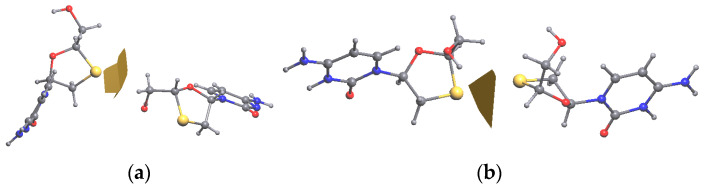
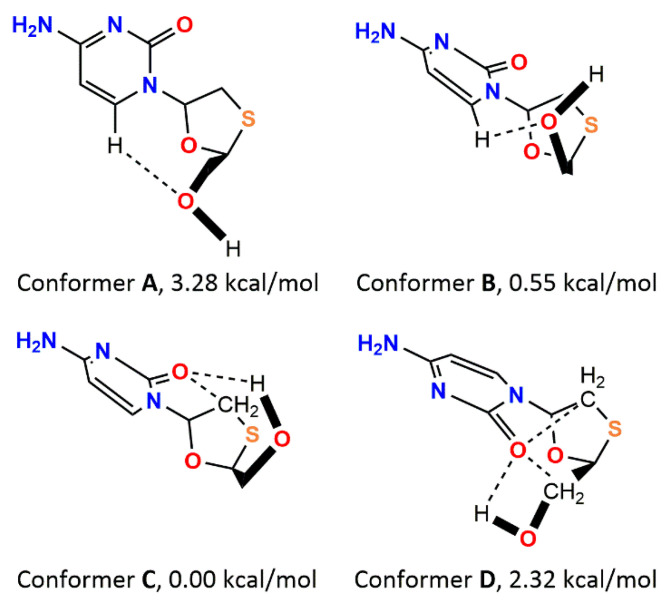
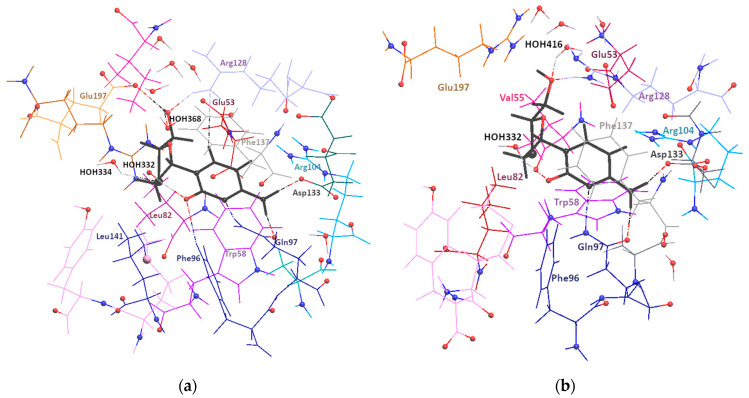
Korlyukov Alexander A, Stash Adam I, Romanenko Alexander R, Trzybiński Damian, Woźniak Krzysztof, Vologzhanina Anna V
A. N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, 28 Vavilov St., Moscow 19334, Russia.
Biological and Chemical Research Centre, Department of Chemistry, University of Warsaw, Żwirki i Wigury 101, 02-089 Warszawa, Poland.
Biomedicines. 2023 Mar 1;11(3):743. doi: 10.3390/biomedicines11030743.
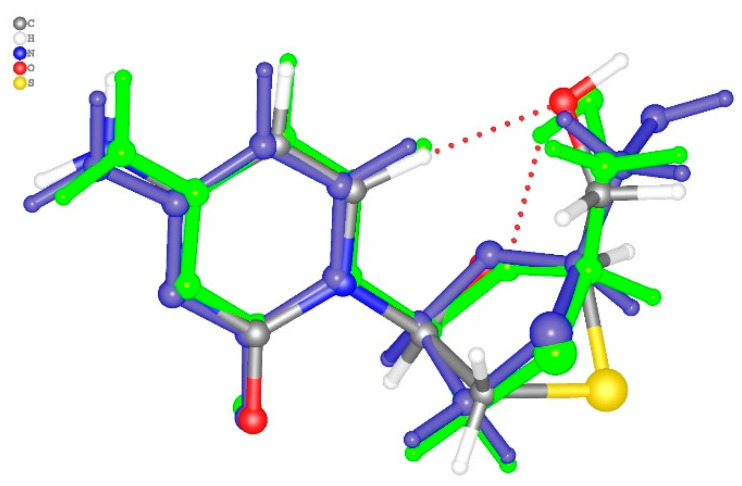
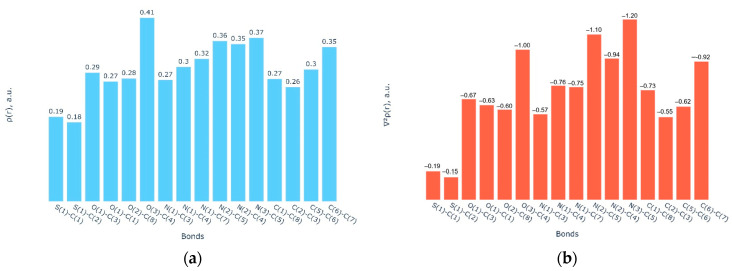
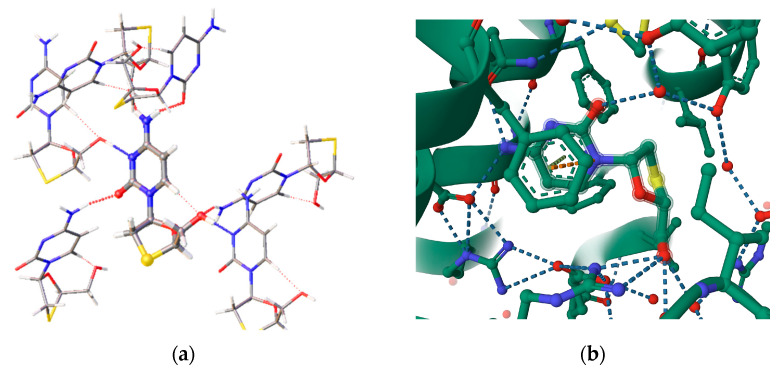
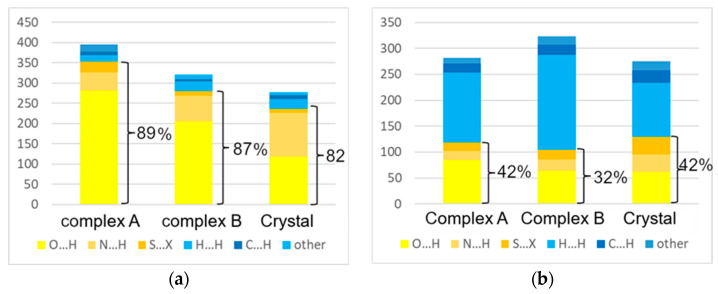
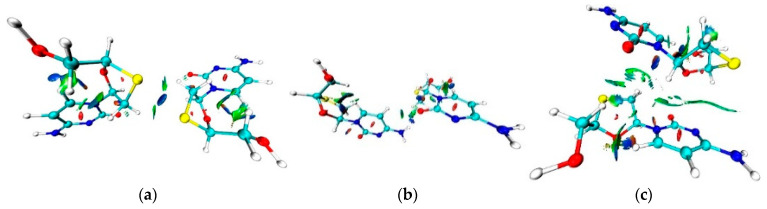
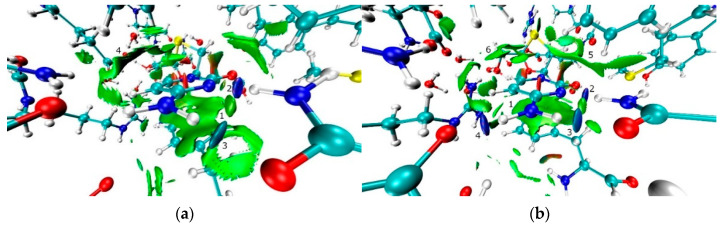
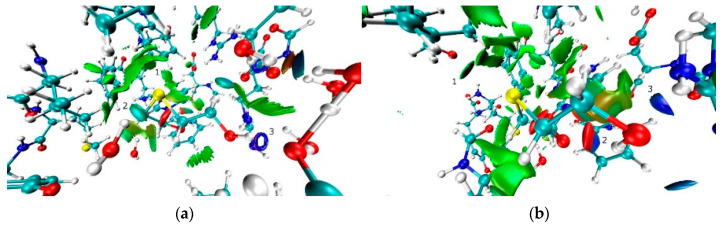
The nature and strength of interactions for an anti-HIV drug, , were studied in a pure crystal form of the drug and the ligand-receptor complexes. High-resolution single-crystal X-ray diffraction studies of the tetragonal polymorph allowed the drug's experimental charge density distribution in the solid state to be obtained. The QM/MM calculations were performed for a simplified model of the complex with deoxycytidine kinase (two complexes with different binding modes) to reconstruct the theoretical charge density distribution. The peculiarities of intramolecular interactions were compared with previously reported data for an isolated molecule. Intermolecular interactions were revealed within the quantum theory of 'Atoms in Molecules', and their contributions to the total crystal energy or ligand-receptor binding energy were evaluated. It was demonstrated that the crystal field effect weakened the intramolecular interactions. Overall, the energies of intermolecular interactions in ligand-receptor complexes (320.1-394.8 kJ/mol) were higher than the energies of interactions in the crystal (276.9 kJ/mol) due to the larger number of hydrophilic interactions. In contrast, the sum of the energies of hydrophobic interactions was found to be unchanged. It was demonstrated by means of the Voronoi tessellation that molecular volume remained constant for different molecular conformations (250(13) Å) and increased up to 399 Å and 521(30) Å for the phosphate and triphosphate.
对一种抗HIV药物(此处药物名称缺失)的相互作用性质和强度,在该药物的纯晶体形式以及配体 - 受体复合物中进行了研究。通过对四方多晶型物的高分辨率单晶X射线衍射研究,获得了该药物在固态下的实验电荷密度分布。针对与脱氧胞苷激酶的复合物简化模型(两种具有不同结合模式的复合物)进行了量子力学/分子力学(QM/MM)计算,以重建理论电荷密度分布。将分子内相互作用的特性与先前报道的孤立分子的数据进行了比较。在“分子中的原子”量子理论范围内揭示了分子间相互作用,并评估了它们对总晶体能量或配体 - 受体结合能的贡献。结果表明,晶体场效应削弱了分子内相互作用。总体而言,由于亲水性相互作用数量较多,配体 - 受体复合物中分子间相互作用的能量(320.1 - 394.8 kJ/mol)高于晶体中相互作用的能量(276.9 kJ/mol)。相比之下,发现疏水相互作用能量的总和保持不变。通过Voronoi镶嵌法表明,对于不同的分子构象,分子体积保持恒定(250(13) Å),而对于磷酸酯和三磷酸酯,分子体积分别增加到399 Å和521(30) Å。