Freda Philip J, Ghosh Attri, Zhang Elizabeth, Luo Tianhao, Chitre Apurva S, Polesskaya Oksana, St Pierre Celine L, Gao Jianjun, Martin Connor D, Chen Hao, Garcia-Martinez Angel G, Wang Tengfei, Han Wenyan, Ishiwari Keita, Meyer Paul, Lamparelli Alexander, King Christopher P, Palmer Abraham A, Li Ruowang, Moore Jason H
Department of Computational Biomedicine, Cedars-Sinai Medical Center, 700 N. San Vicente Blvd., Pacific Design Center, Suite G540, West Hollywood, CA, 90069, USA.
Department of Psychiatry, University of California San Diego, 9500 Gilman Dr., Mail Code: 0667, La Jolla, CA, 92093-0667, USA.
BioData Min. 2023 Apr 10;16(1):14. doi: 10.1186/s13040-023-00331-3.
Quantitative Trait Locus (QTL) analysis and Genome-Wide Association Studies (GWAS) have the power to identify variants that capture significant levels of phenotypic variance in complex traits. However, effort and time are required to select the best methods and optimize parameters and pre-processing steps. Although machine learning approaches have been shown to greatly assist in optimization and data processing, applying them to QTL analysis and GWAS is challenging due to the complexity of large, heterogenous datasets. Here, we describe proof-of-concept for an automated machine learning approach, AutoQTL, with the ability to automate many complicated decisions related to analysis of complex traits and generate solutions to describe relationships that exist in genetic data.
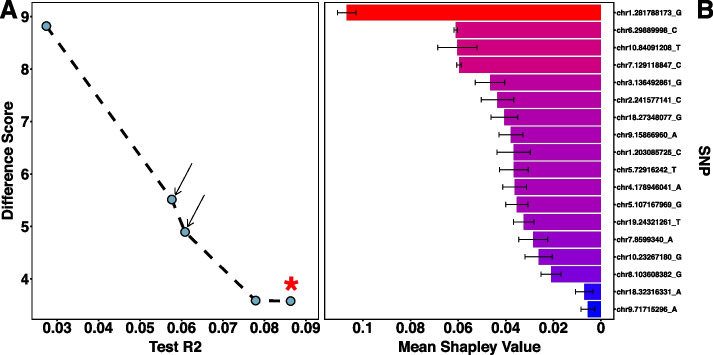
Using a publicly available dataset of 18 putative QTL from a large-scale GWAS of body mass index in the laboratory rat, Rattus norvegicus, AutoQTL captures the phenotypic variance explained under a standard additive model. AutoQTL also detects evidence of non-additive effects including deviations from additivity and 2-way epistatic interactions in simulated data via multiple optimal solutions. Additionally, feature importance metrics provide different insights into the inheritance models and predictive power of multiple GWAS-derived putative QTL.
This proof-of-concept illustrates that automated machine learning techniques can complement standard approaches and have the potential to detect both additive and non-additive effects via various optimal solutions and feature importance metrics. In the future, we aim to expand AutoQTL to accommodate omics-level datasets with intelligent feature selection and feature engineering strategies.
数量性状基因座(QTL)分析和全基因组关联研究(GWAS)有能力识别在复杂性状中捕获显著表型变异水平的变异。然而,选择最佳方法、优化参数和预处理步骤需要付出努力和时间。尽管机器学习方法已被证明能极大地协助优化和数据处理,但由于大型异质数据集的复杂性,将其应用于QTL分析和GWAS具有挑战性。在这里,我们描述了一种自动化机器学习方法AutoQTL的概念验证,它能够自动做出许多与复杂性状分析相关的复杂决策,并生成解决方案来描述遗传数据中存在的关系。
使用来自实验室大鼠褐家鼠体重指数大规模GWAS的18个假定QTL的公开可用数据集,AutoQTL捕获了标准加性模型下解释的表型变异。AutoQTL还通过多个最优解检测到非加性效应的证据,包括模拟数据中与加性的偏差和双向上位性相互作用。此外,特征重要性指标为多个GWAS衍生的假定QTL的遗传模型和预测能力提供了不同的见解。
这一概念验证表明,自动化机器学习技术可以补充标准方法,并有潜力通过各种最优解和特征重要性指标检测加性和非加性效应。未来,我们旨在扩展AutoQTL,以通过智能特征选择和特征工程策略来适应组学水平的数据集。