Becker Devan, Champredon David, Chato Connor, Gugan Gopi, Poon Art
Department of Pathology and Laboratory Medicine, Schulich School of Medicine and Dentistry, Western University, London, Ontario, Canada.
Public Health Agency of Canada, National Microbiology Laboratory, Public Health Risk Sciences Division, Guelph, Ontario, Canada.
NAR Genom Bioinform. 2023 Apr 24;5(2):lqad038. doi: 10.1093/nargab/lqad038. eCollection 2023 Jun.
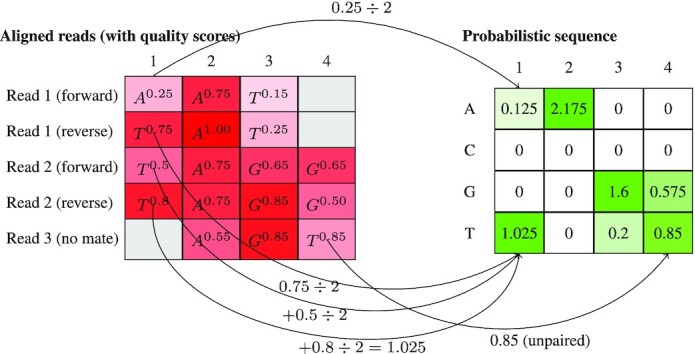
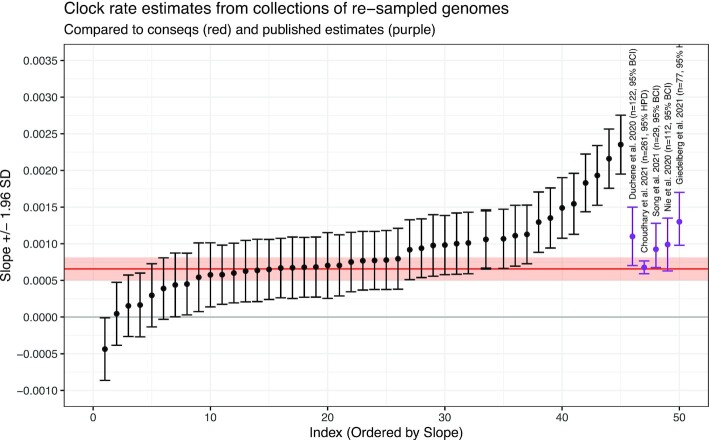
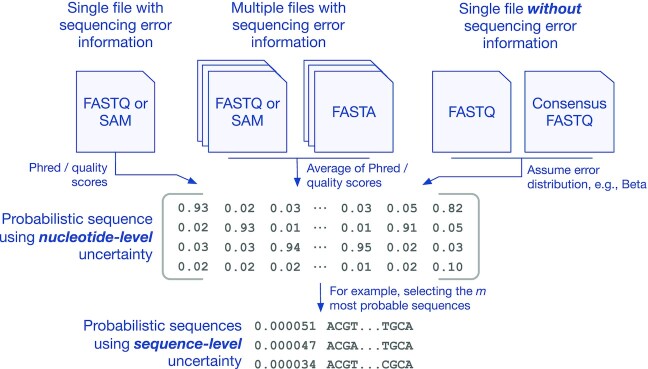
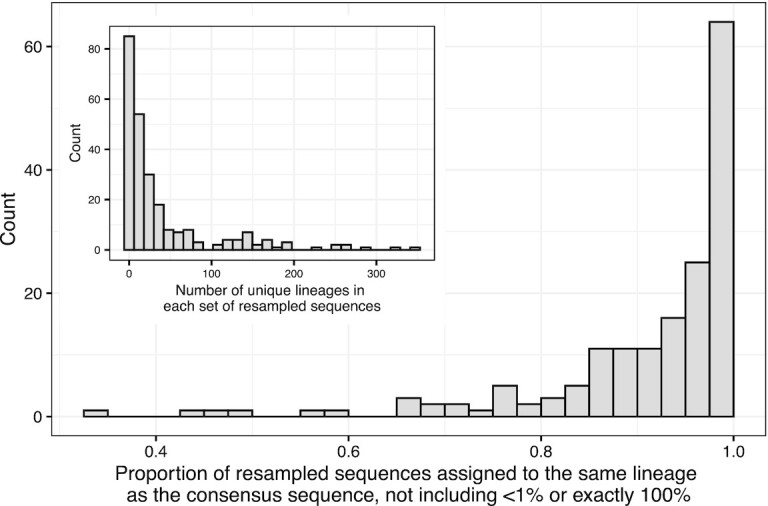
Genetic sequencing is subject to many different types of errors, but most analyses treat the resultant sequences as if they are known without error. Next generation sequencing methods rely on significantly larger numbers of reads than previous sequencing methods in exchange for a loss of accuracy in each individual read. Still, the coverage of such machines is imperfect and leaves uncertainty in many of the base calls. In this work, we demonstrate that the uncertainty in sequencing techniques will affect downstream analysis and propose a straightforward method to propagate the uncertainty. Our method (which we have dubbed Sequence Uncertainty Propagation, or SUP) uses a probabilistic matrix representation of individual sequences which incorporates base quality scores as a measure of uncertainty that naturally lead to resampling and replication as a framework for uncertainty propagation. With the matrix representation, resampling possible base calls according to quality scores provides a bootstrap- or prior distribution-like first step towards genetic analysis. Analyses based on these re-sampled sequences will include a more complete evaluation of the error involved in such analyses. We demonstrate our resampling method on SARS-CoV-2 data. The resampling procedures add a linear computational cost to the analyses, but the large impact on the variance in downstream estimates makes it clear that ignoring this uncertainty may lead to overly confident conclusions. We show that SARS-CoV-2 lineage designations via Pangolin are much less certain than the bootstrap support reported by Pangolin would imply and the clock rate estimates for SARS-CoV-2 are much more variable than reported.
基因测序容易出现多种不同类型的错误,但大多数分析都将所得序列视为没有错误的已知序列。与之前的测序方法相比,新一代测序方法依赖大量的读数,以换取单个读数准确性的降低。尽管如此,此类机器的覆盖并不完美,在许多碱基识别中仍存在不确定性。在这项工作中,我们证明了测序技术中的不确定性会影响下游分析,并提出了一种简单的方法来传播这种不确定性。我们的方法(我们称之为序列不确定性传播,即SUP)使用单个序列的概率矩阵表示,将碱基质量分数纳入其中,作为不确定性的一种度量,这自然会导致重采样和复制,作为不确定性传播的框架。通过矩阵表示,根据质量分数对可能的碱基识别进行重采样,为基因分析提供了类似自展或先验分布的第一步。基于这些重采样序列的分析将对这类分析中涉及的误差进行更全面的评估。我们在新冠病毒数据上展示了我们的重采样方法。重采样过程给分析增加了线性计算成本,但对下游估计方差的巨大影响表明,忽略这种不确定性可能会导致过于自信的结论。我们表明,通过穿山甲软件进行的新冠病毒谱系分类比穿山甲软件报告的自展支持所暗示的确定性要低得多,而且新冠病毒的时钟速率估计比报告的更具变异性。