Geidelberg Lily, Boyd Olivia, Jorgensen David, Siveroni Igor, Nascimento Fabrícia F, Johnson Robert, Ragonnet-Cronin Manon, Fu Han, Wang Haowei, Xi Xiaoyue, Chen Wei, Liu Dehui, Chen Yingying, Tian Mengmeng, Tan Wei, Zai Junjie, Sun Wanying, Li Jiandong, Li Junhua, Volz Erik M, Li Xingguang, Nie Qing
Department of Infectious Disease Epidemiology and MRC Centre for Global Infectious Disease Analysis, Imperial College London, Norfolk Place W2 1PG, UK.
Department of Mathematics, Imperial College London, London SW7 2AZ, UK.
Virus Evol. 2021 Mar 14;7(1):veaa102. doi: 10.1093/ve/veaa102. eCollection 2021 Jan.
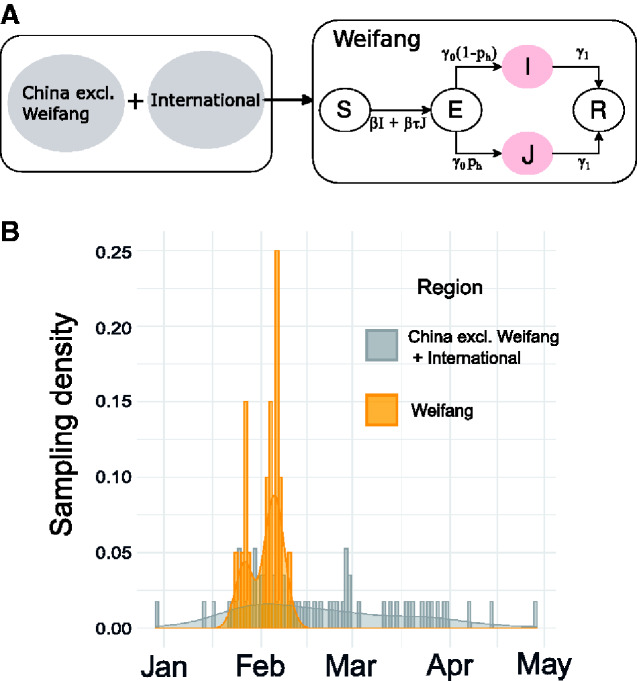
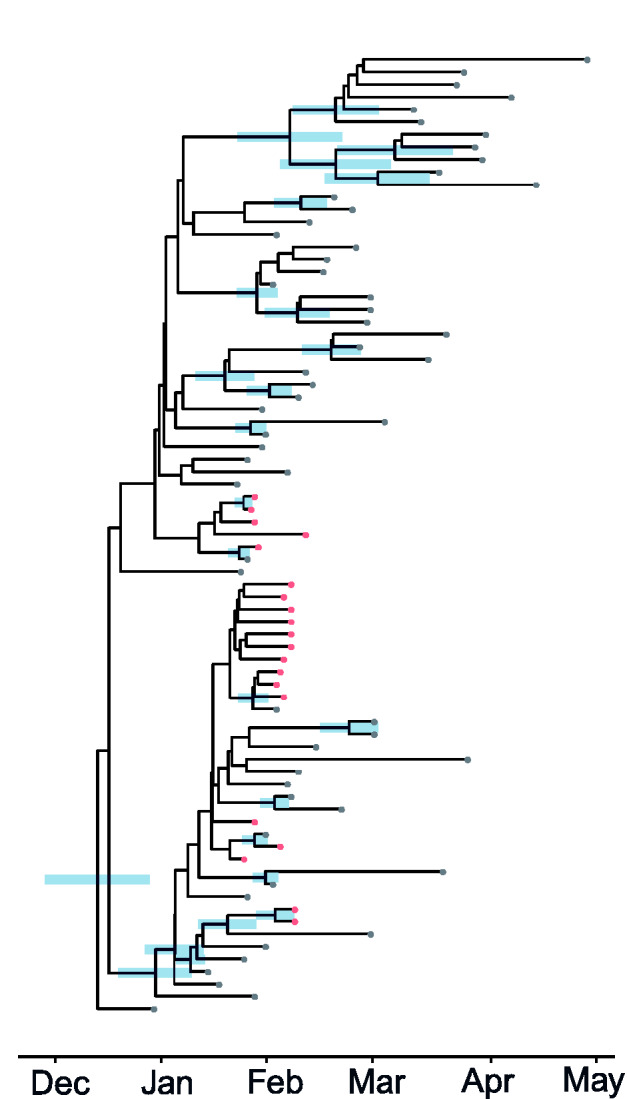
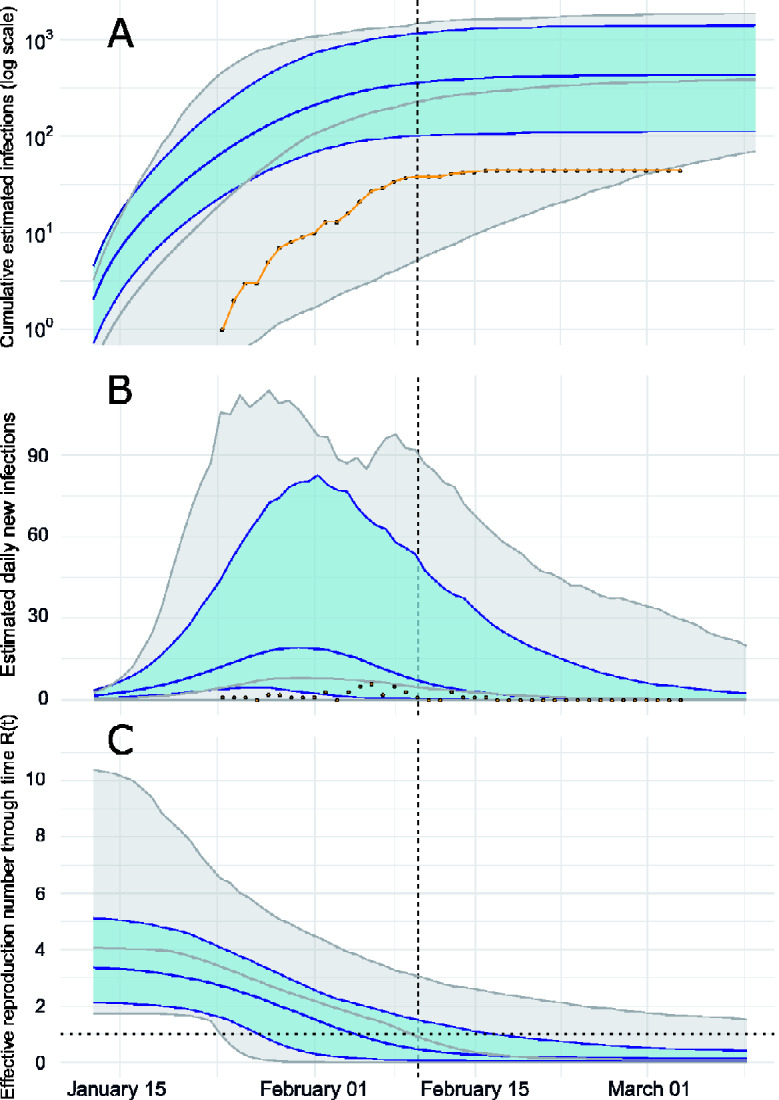
Analysis of genetic sequence data from the SARS-CoV-2 pandemic can provide insights into epidemic origins, worldwide dispersal, and epidemiological history. With few exceptions, genomic epidemiological analysis has focused on geographically distributed data sets with few isolates in any given location. Here, we report an analysis of 20 whole SARS- CoV-2 genomes from a single relatively small and geographically constrained outbreak in Weifang, People's Republic of China. Using Bayesian model-based phylodynamic methods, we estimate a mean basic reproduction number ( ) of 3.4 (95% highest posterior density interval: 2.1-5.2) in Weifang, and a mean effective reproduction number ( that falls below 1 on 4 February. We further estimate the number of infections through time and compare these estimates to confirmed diagnoses by the Weifang Centers for Disease Control. We find that these estimates are consistent with reported cases and there is unlikely to be a large undiagnosed burden of infection over the period we studied.
对严重急性呼吸综合征冠状病毒2(SARS-CoV-2)大流行的基因序列数据进行分析,有助于深入了解疫情起源、全球传播情况及流行病学历史。除少数例外情况,基因组流行病学分析主要集中在地理分布数据集上,在任何给定地点的分离株数量都很少。在此,我们报告了对来自中国山东省潍坊市一次相对较小且地理范围有限的疫情中的20个完整SARS-CoV-2基因组的分析。使用基于贝叶斯模型的系统发育动力学方法,我们估计潍坊市的平均基本再生数(R0)为3.4(95%最高后验密度区间:2.1 - 5.2),平均有效再生数(Rt)在2月4日降至1以下。我们还进一步估计了随时间推移的感染数量,并将这些估计值与潍坊市疾病预防控制中心的确诊病例进行比较。我们发现这些估计值与报告病例一致,并且在我们研究的时间段内,不太可能存在大量未被诊断的感染负担。