Department of Materials Engineering, The University of Tokyo, Tokyo 113-0033, Japan.
Institute of Industrial Science, The University of Tokyo, Tokyo 113-0033, Japan.
J Phys Chem Lett. 2023 May 25;14(20):4858-4865. doi: 10.1021/acs.jpclett.3c00142. Epub 2023 May 18.
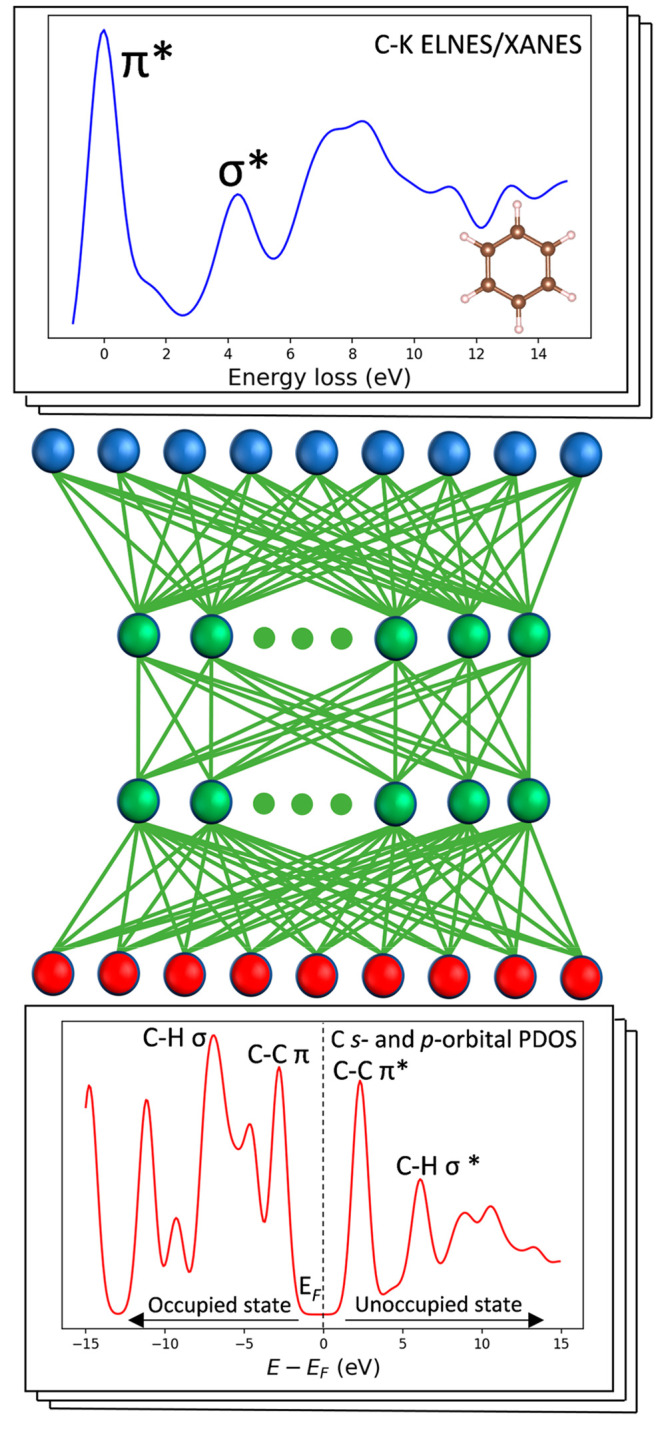
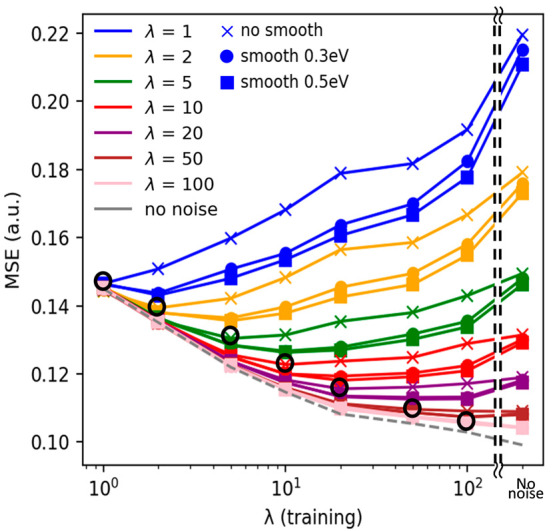
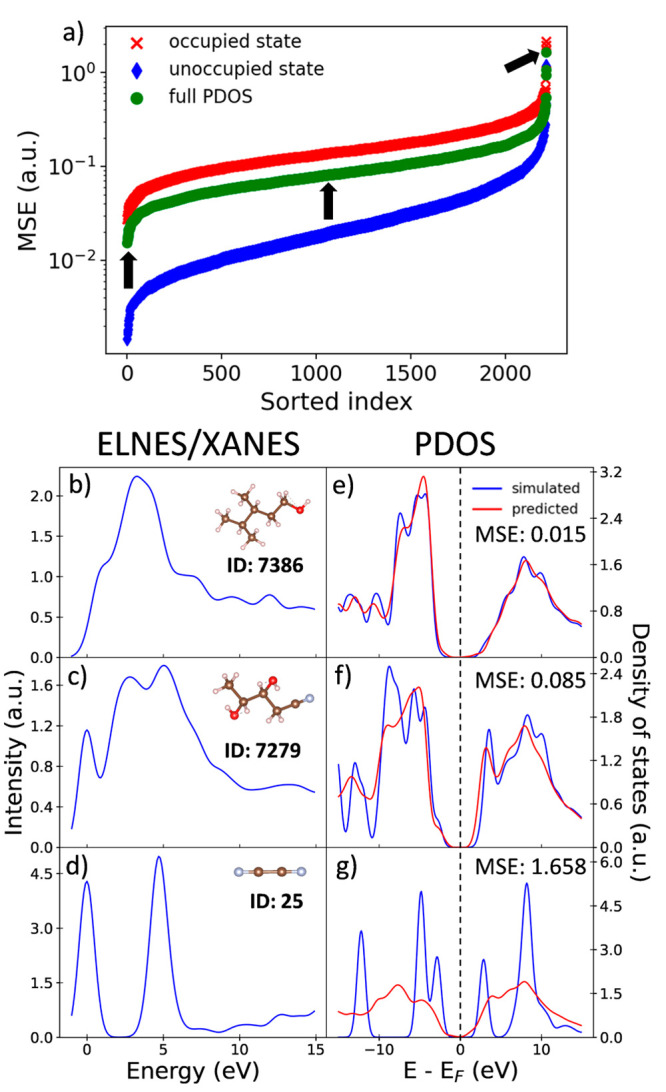
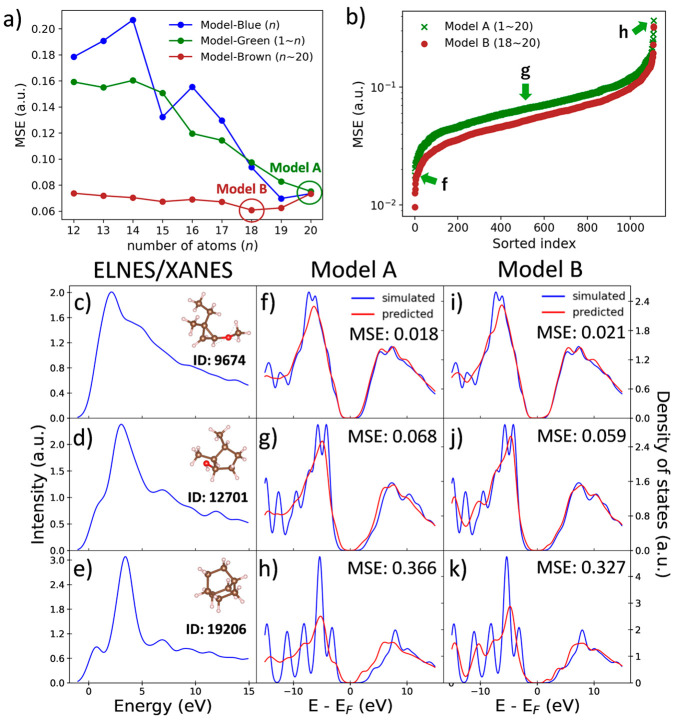
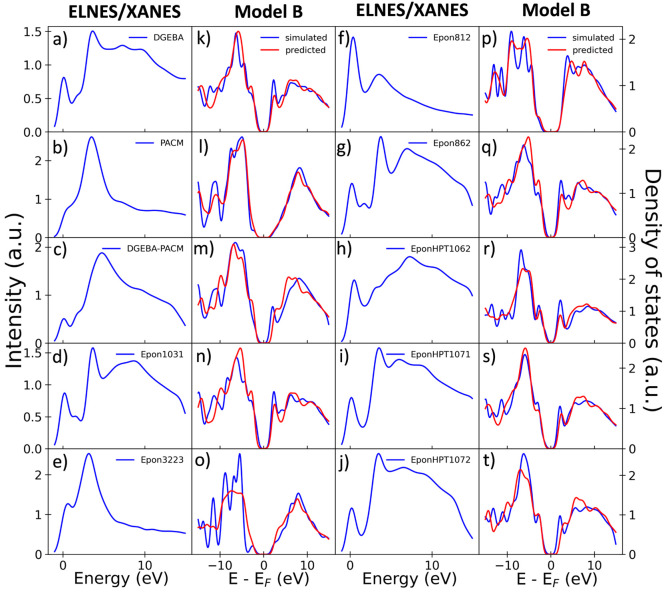
The core-loss spectrum reflects the partial density of states (PDOS) of the unoccupied states at the excited state and is a powerful analytical technique to investigate local atomic and electronic structures of materials. However, various molecular properties governed by the ground-state electronic structure of the occupied orbital cannot be directly obtained from the core-loss spectra. Here, we constructed a machine learning model to predict the ground-state carbon s- and p-orbital PDOS in both occupied and unoccupied states from the C K-edge spectra. We also attempted an extrapolation prediction of the PDOS of larger molecules using a model trained by smaller molecules and found that the extrapolation prediction performance can be improved by excluding tiny molecules. Besides, we found that using smoothing preprocess and training by specific noise data can improve the PDOS prediction for noise-contained spectra, which pave a way for the application of the prediction model to the experimental data.
核心损耗谱反映了激发态下未占据态的部分态密度(PDOS),是研究材料局部原子和电子结构的强大分析技术。然而,由占据轨道的基态电子结构决定的各种分子性质不能直接从核心损耗谱中获得。在这里,我们构建了一个机器学习模型,从 C K 边缘谱中预测占据和未占据态下的碳 s 和 p 轨道 PDOS。我们还尝试使用小分子训练的模型对更大分子的 PDOS 进行外推预测,发现通过排除微小分子可以提高外推预测性能。此外,我们发现使用平滑预处理和特定噪声数据训练可以提高包含噪声谱的 PDOS 预测,为将预测模型应用于实验数据铺平了道路。