Luxembourg Centre for Systems Biomedicine (LCSB), University of Luxembourg, Avenue du Swing 6, Belvaux, L-4367, Luxembourg.
Anal Bioanal Chem. 2023 Jul;415(17):3415-3434. doi: 10.1007/s00216-023-04724-5. Epub 2023 May 22.
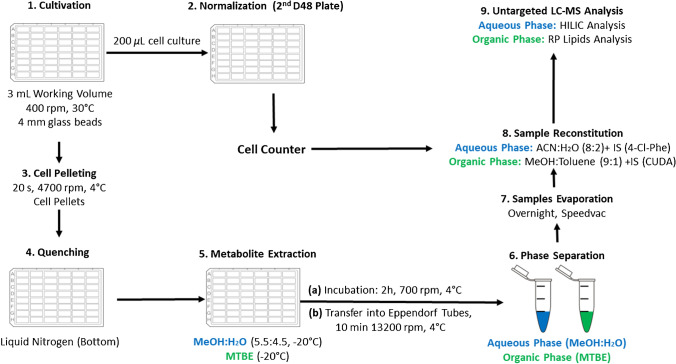
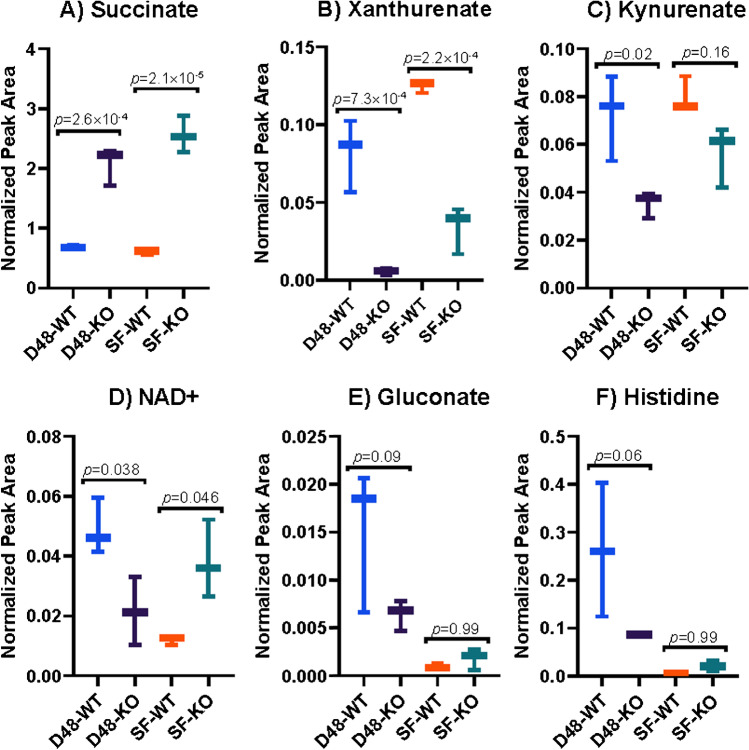
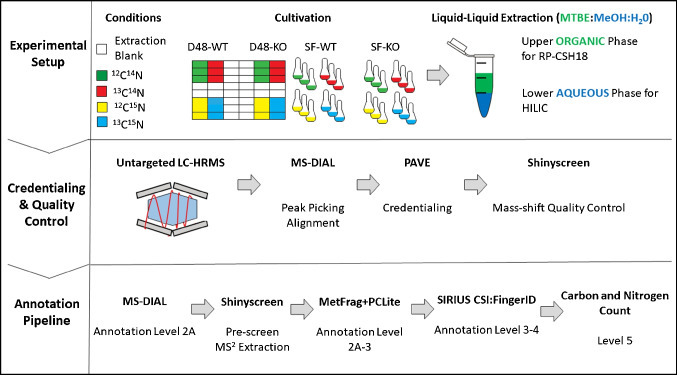
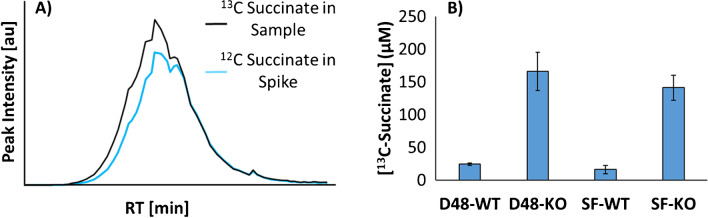
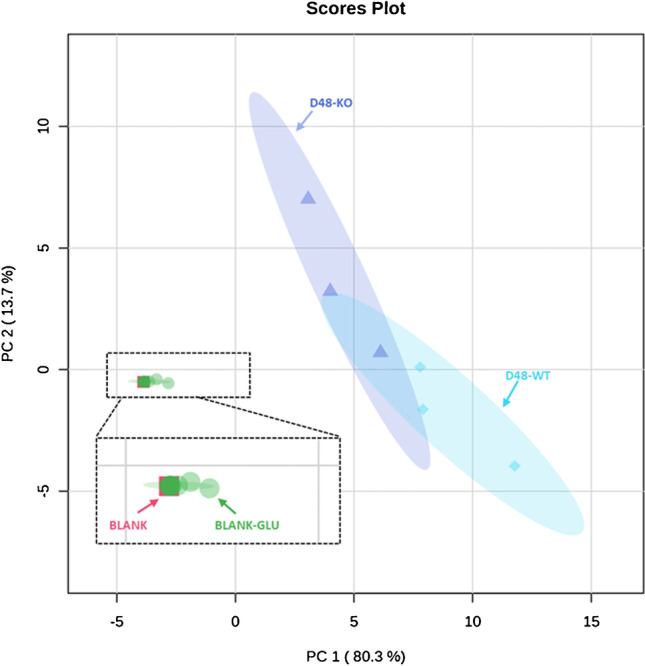
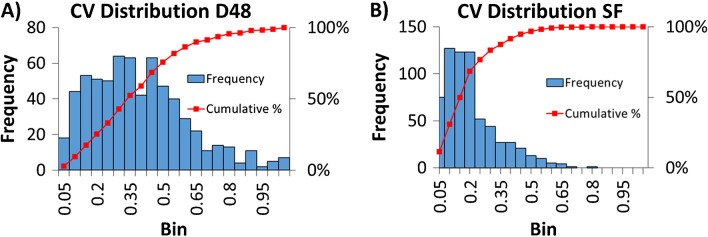
Identifying metabolites in model organisms is critical for many areas of biology, including unravelling disease aetiology or elucidating functions of putative enzymes. Even now, hundreds of predicted metabolic genes in Saccharomyces cerevisiae remain uncharacterized, indicating that our understanding of metabolism is far from complete even in well-characterized organisms. While untargeted high-resolution mass spectrometry (HRMS) enables the detection of thousands of features per analysis, many of these have a non-biological origin. Stable isotope labelling (SIL) approaches can serve as credentialing strategies to distinguish biologically relevant features from background signals, but implementing these experiments at large scale remains challenging. Here, we developed a SIL-based approach for high-throughput untargeted metabolomics in S. cerevisiae, including deep-48 well format-based cultivation and metabolite extraction, building on the peak annotation and verification engine (PAVE) tool. Aqueous and nonpolar extracts were analysed using HILIC and RP liquid chromatography, respectively, coupled to Orbitrap Q Exactive HF mass spectrometry. Of the approximately 37,000 total detected features, only 3-7% of the features were credentialed and used for data analysis with open-source software such as MS-DIAL, MetFrag, Shinyscreen, SIRIUS CSI:FingerID, and MetaboAnalyst, leading to the successful annotation of 198 metabolites using MS database matching. Comparable metabolic profiles were observed for wild-type and sdh1Δ yeast strains grown in deep-48 well plates versus the classical shake flask format, including the expected increase in intracellular succinate concentration in the sdh1Δ strain. The described approach enables high-throughput yeast cultivation and credentialing-based untargeted metabolomics, providing a means to efficiently perform molecular phenotypic screens and help complete metabolic networks.
鉴定模式生物中的代谢物对于生物学的许多领域都至关重要,包括揭示疾病病因或阐明假定酶的功能。即使在现在,酿酒酵母中仍有数百个预测的代谢基因尚未被描述,这表明即使在研究透彻的生物体中,我们对代谢的理解也远未完成。虽然非靶向高分辨率质谱(HRMS)能够在每次分析中检测到数千个特征,但其中许多都具有非生物起源。稳定同位素标记(SIL)方法可以作为认证策略,将生物学上相关的特征与背景信号区分开来,但在大规模实施这些实验仍然具有挑战性。在这里,我们开发了一种基于 SIL 的酿酒酵母高通量非靶向代谢组学方法,包括基于深 48 孔板格式的培养和代谢物提取,该方法基于峰注释和验证引擎(PAVE)工具。分别使用亲水相互作用色谱(HILIC)和反相色谱(RP)对水相和非极性提取物进行分析,分别与轨道阱 Q Exactive HF 质谱仪联用。在大约 37000 个总检测到的特征中,只有 3-7%的特征被认证并用于使用开源软件(如 MS-DIAL、MetFrag、Shinyscreen、SIRIUS CSI:FingerID 和 MetaboAnalyst)进行数据分析,从而成功地使用 MS 数据库匹配注释了 198 种代谢物。在深 48 孔板和经典摇瓶培养中,野生型和 sdh1Δ酵母菌株的代谢谱相似,包括 sdh1Δ 菌株中细胞内琥珀酸浓度的预期增加。所描述的方法能够实现高通量酵母培养和基于认证的非靶向代谢组学,为有效地进行分子表型筛选提供了一种手段,并有助于完成代谢网络。