Lewis Sigler Institute for Integrative Genomics , Princeton University , Princeton , New Jersey 08544 , United States.
Department of Chemistry , Princeton University , Princeton , New Jersey 08544 , United States.
Anal Chem. 2019 Feb 5;91(3):1838-1846. doi: 10.1021/acs.analchem.8b03132. Epub 2019 Jan 10.
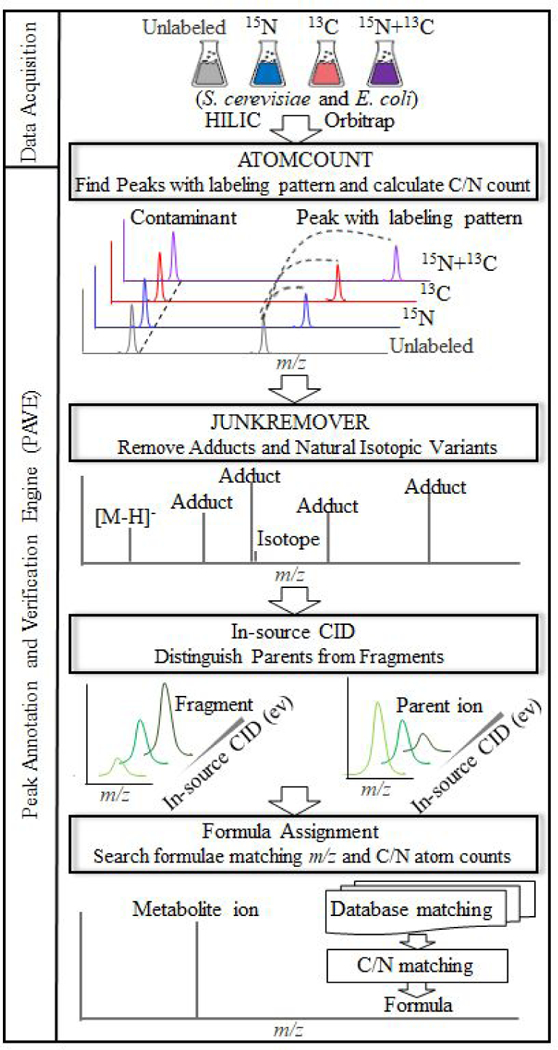
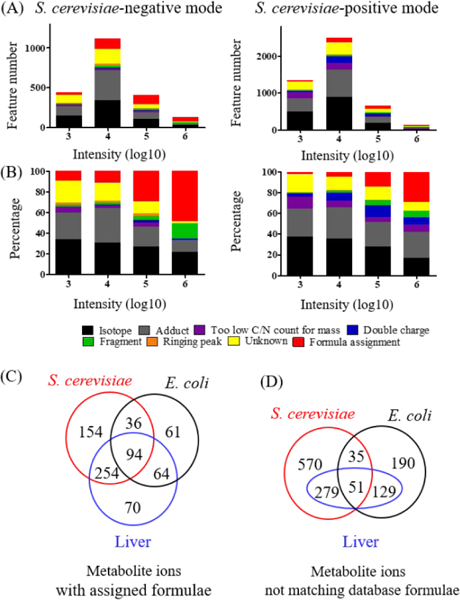
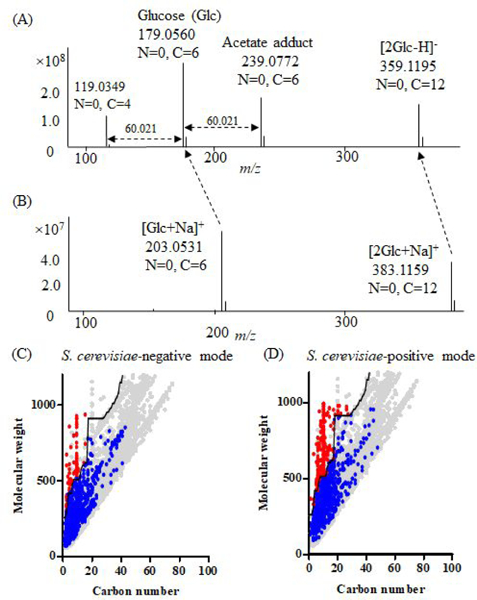
Untargeted metabolomics can detect more than 10 000 peaks in a single LC-MS run. The correspondence between these peaks and metabolites, however, remains unclear. Here, we introduce a Peak Annotation and Verification Engine (PAVE) for annotating untargeted microbial metabolomics data. The workflow involves growing cells in C and N isotope-labeled media to identify peaks from biological compounds and their carbon and nitrogen atom counts. Improved deisotoping and deadducting are enabled by algorithms that integrate positive mode, negative mode, and labeling data. To distinguish metabolites and their fragments, PAVE experimentally measures the response of each peak to weak in-source collision induced dissociation, which increases the peak intensity for fragments while decreasing it for their parent ions. The molecular formulas of the putative metabolites are then assigned based on database searching using both m/ z and C/N atom counts. Application of this procedure to Saccharomyces cerevisiae and Escherichia coli revealed that more than 80% of peaks do not label, i.e., are environmental contaminants. More than 70% of the biological peaks are isotopic variants, adducts, fragments, or mass spectrometry artifacts yielding ∼2000 apparent metabolites across the two organisms. About 650 match to a known metabolite formula based on m/ z and C/N atom counts, with 220 assigned structures based on MS/MS and/or retention time to match to authenticated standards. Thus, PAVE enables systematic annotation of LC-MS metabolomics data with only ∼4% of peaks annotated as apparent metabolites.
非靶向代谢组学可以在单次 LC-MS 运行中检测到超过 10000 个峰。然而,这些峰与代谢物之间的对应关系尚不清楚。在这里,我们介绍了一种用于注释非靶向微生物代谢组学数据的峰注释和验证引擎(PAVE)。该工作流程涉及在 C 和 N 同位素标记的培养基中培养细胞,以鉴定来自生物化合物及其碳和氮原子数的峰。通过整合正模式、负模式和标记数据的算法,实现了改进的去同位素化和去加成。为了区分代谢物及其片段,PAVE 通过实验测量每个峰对弱源内碰撞诱导解离的响应,从而增加片段的峰强度,同时降低其母体离子的峰强度。然后根据数据库搜索,使用 m/z 和 C/N 原子数对假定代谢物的分子式进行分配。将该程序应用于酿酒酵母和大肠杆菌,结果表明,超过 80%的峰不标记,即属于环境污染物。超过 70%的生物峰是同位素变体、加合物、片段或质谱伪影,这两种生物产生的表观代谢物约有 2000 种。约有 650 种根据 m/z 和 C/N 原子数匹配到已知代谢物公式,其中 220 种根据 MS/MS 和/或保留时间分配结构以匹配经认证的标准。因此,PAVE 可以对 LC-MS 代谢组学数据进行系统注释,只有约 4%的峰被注释为表观代谢物。