Sánchez-Serrano Andrea, Mejía Lorena, Camaró Maria Luisa, Ortolá-Malvar Susana, Llácer-Luna Martín, García-González Neris, González-Candelas Fernando
Joint Research Unit "Infection and Public Health", FISABIO-University of Valencia, 46020 Valencia, Spain.
Institute for Integrative Systems Biology (I2SysBio), CSIC-University of Valencia, 46980 Valencia, Spain.
Antibiotics (Basel). 2023 May 9;12(5):883. doi: 10.3390/antibiotics12050883.
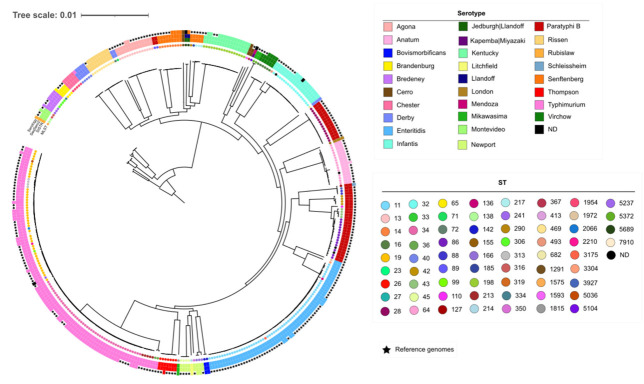
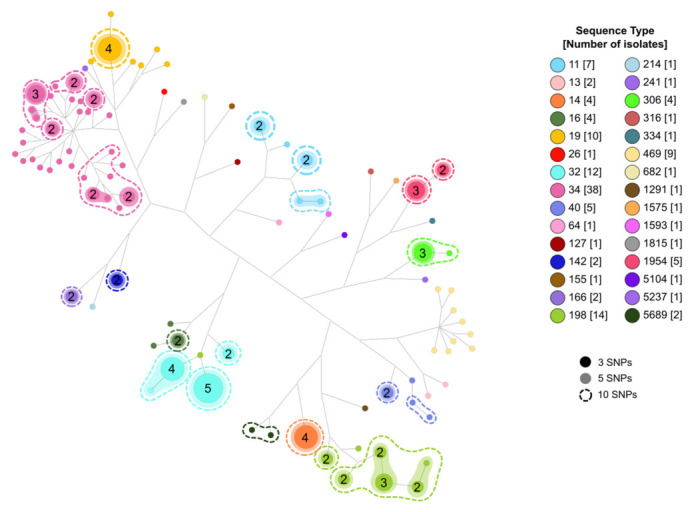
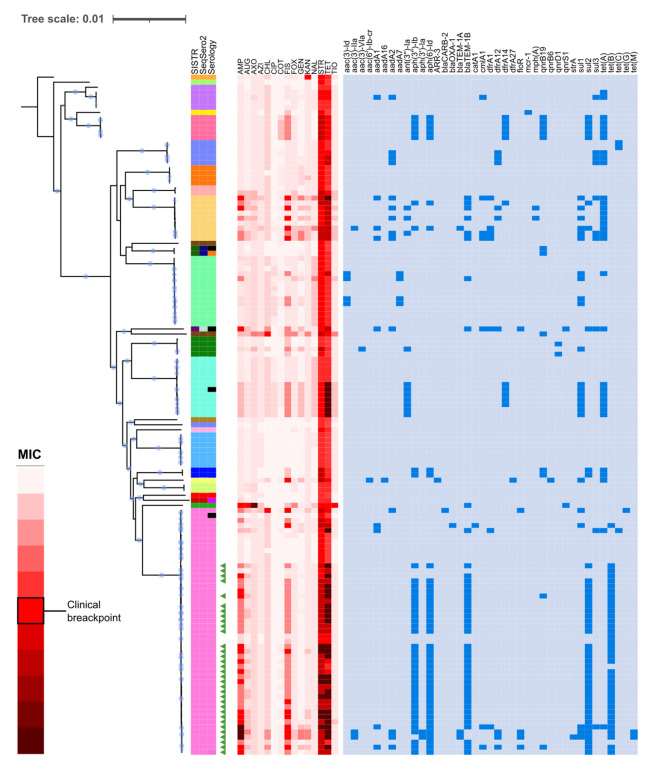
subspecies is one of the most important foodborne pathogens and the causative agent of salmonellosis, which affects both humans and animals producing numerous infections every year. The study and understanding of its epidemiology are key to monitoring and controlling these bacteria. With the development of whole-genome sequencing (WGS) technologies, surveillance based on traditional serotyping and phenotypic tests of resistance is being replaced by genomic surveillance. To introduce WGS as a routine methodology for the surveillance of food-borne in the region, we applied this technology to analyze a set of 141 isolates obtained from various food sources between 2010 and 2017 in the Comunitat Valenciana (Spain). For this, we performed an evaluation of the most relevant typing methods, serotyping and sequence typing, using both traditional and in silico approaches. We extended the use of WGS to detect antimicrobial resistance determinants and predicted minimum inhibitory concentrations (MICs). Finally, to understand possible contaminant sources in this region and their relationship to antimicrobial resistance (AMR), we performed cluster detection combining single-nucleotide polymorphism (SNP) pairwise distances and phylogenetic and epidemiological data. The results of in silico serotyping with WGS data were highly congruent with those of serological analyses (98.5% concordance). Multi-locus sequence typing (MLST) profiles obtained with WGS information were also highly congruent with the sequence type (ST) assignment based on Sanger sequencing (91.9% coincidence). In silico identification of antimicrobial resistance determinants and minimum inhibitory concentrations revealed a high number of resistance genes and possible resistant isolates. A combined phylogenetic and epidemiological analysis with complete genome sequences revealed relationships among isolates indicative of possible common sources for isolates with separate sampling in time and space that had not been detected from epidemiological information. As a result, we demonstrate the usefulness of WGS and in silico methods to obtain an improved characterization of isolates, allowing better surveillance of the pathogen in food products and in potential environmental and clinical samples of related interest.
亚种是最重要的食源性病原体之一,也是沙门氏菌病的病原体,每年都会影响人类和动物,导致大量感染。对其流行病学的研究和理解是监测和控制这些细菌的关键。随着全基因组测序(WGS)技术的发展,基于传统血清分型和耐药表型测试的监测正被基因组监测所取代。为了将WGS作为该地区食源性病原体监测的常规方法引入,我们应用该技术分析了2010年至2017年间从西班牙巴伦西亚自治区不同食物来源获得的141株分离株。为此,我们使用传统方法和计算机方法对最相关的分型方法(血清分型和序列分型)进行了评估。我们扩展了WGS的应用,以检测抗菌药物耐药决定因素并预测最低抑菌浓度(MIC)。最后,为了了解该地区可能的污染源及其与抗菌药物耐药性(AMR)的关系,我们结合单核苷酸多态性(SNP)成对距离以及系统发育和流行病学数据进行了聚类检测。利用WGS数据进行的计算机血清分型结果与血清学分析结果高度一致(一致性为98.5%)。通过WGS信息获得的多位点序列分型(MLST)图谱也与基于桑格测序的序列类型(ST)分配高度一致(一致性为91.9%)。计算机对抗菌药物耐药决定因素和最低抑菌浓度的鉴定揭示了大量耐药基因和可能的耐药分离株。对完整基因组序列进行的系统发育和流行病学综合分析揭示了分离株之间的关系,表明在时间和空间上单独采样的分离株可能存在共同来源,而这些在流行病学信息中并未检测到。因此,我们证明了WGS和计算机方法在更好地表征分离株方面的有用性,从而能够更好地监测食品以及相关潜在环境和临床样本中的病原体。