Carroll Laura M, Buehler Ariel J, Gaballa Ahmed, Siler Julie D, Cummings Kevin J, Cheng Rachel A, Wiedmann Martin
Department of Food Science, Cornell University, Ithaca, NY, United States.
Department of Population Medicine and Diagnostic Sciences, Cornell University, Ithaca, NY, United States.
Front Microbiol. 2021 Oct 18;12:763669. doi: 10.3389/fmicb.2021.763669. eCollection 2021.
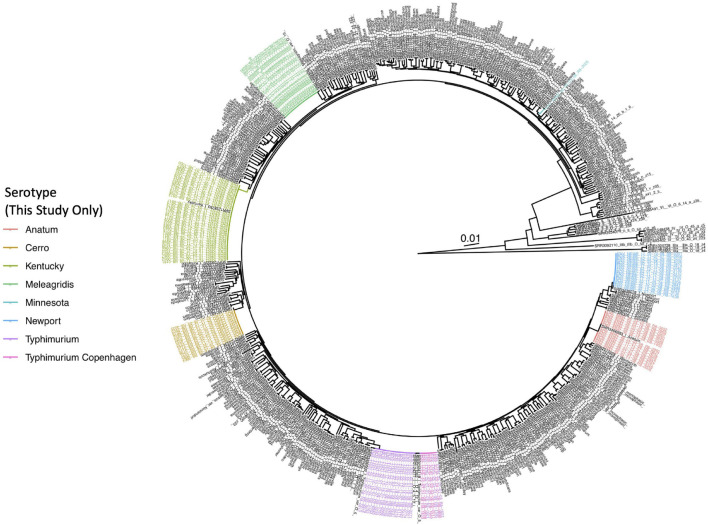
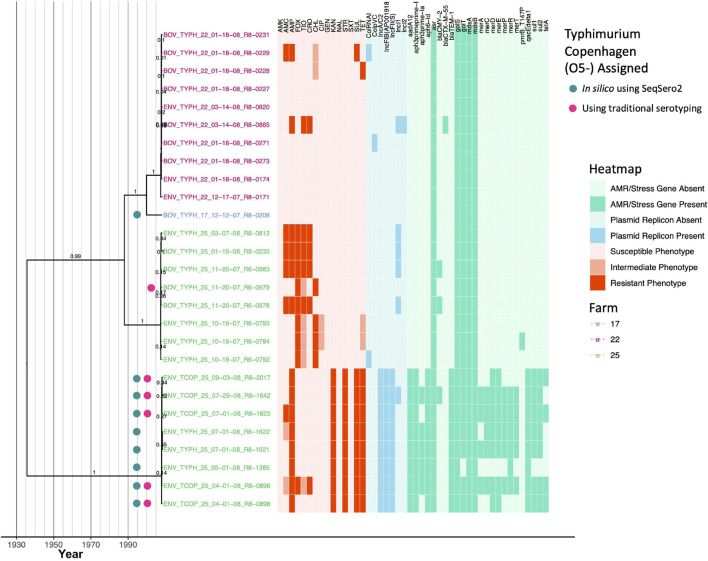
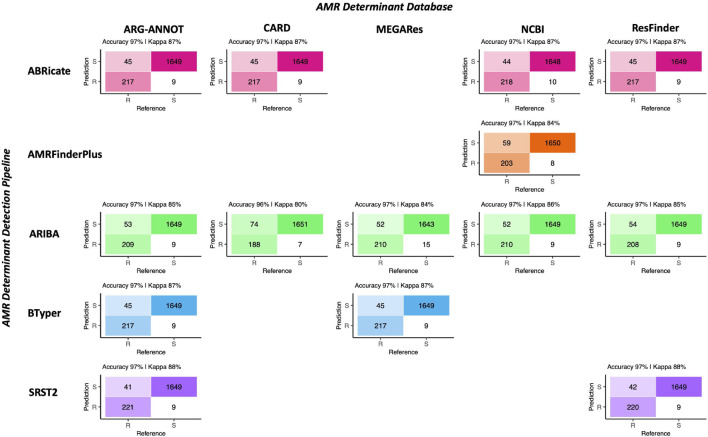
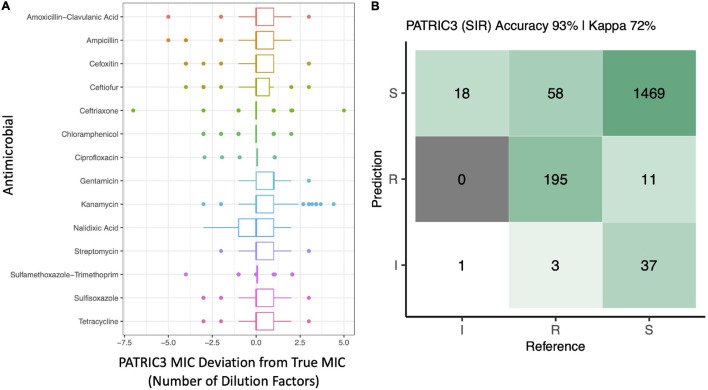
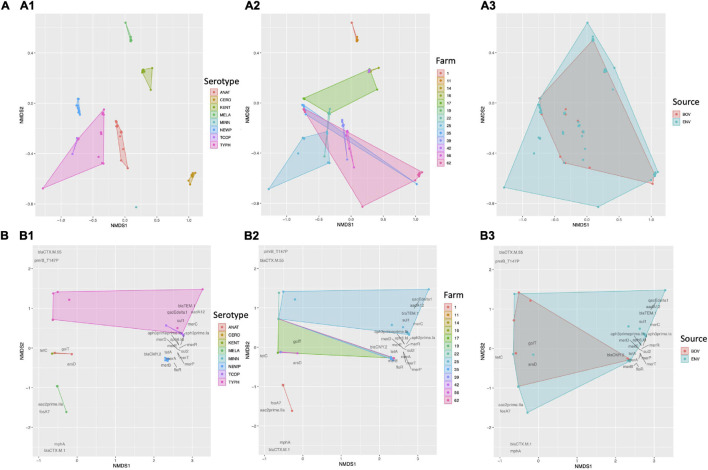
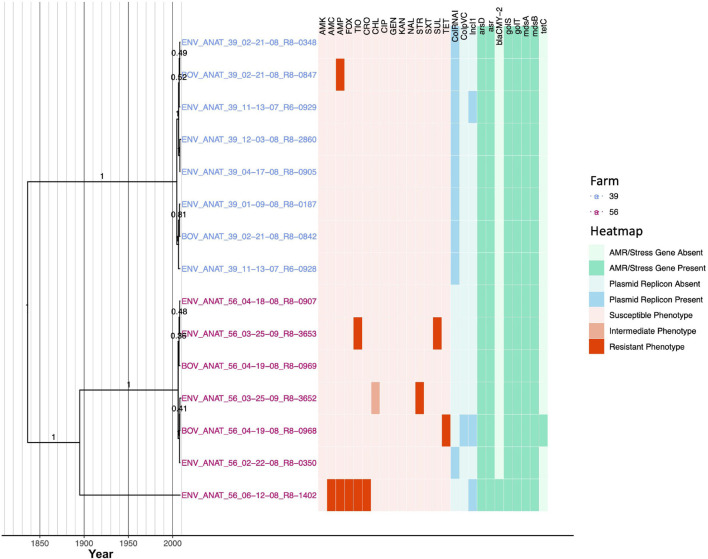
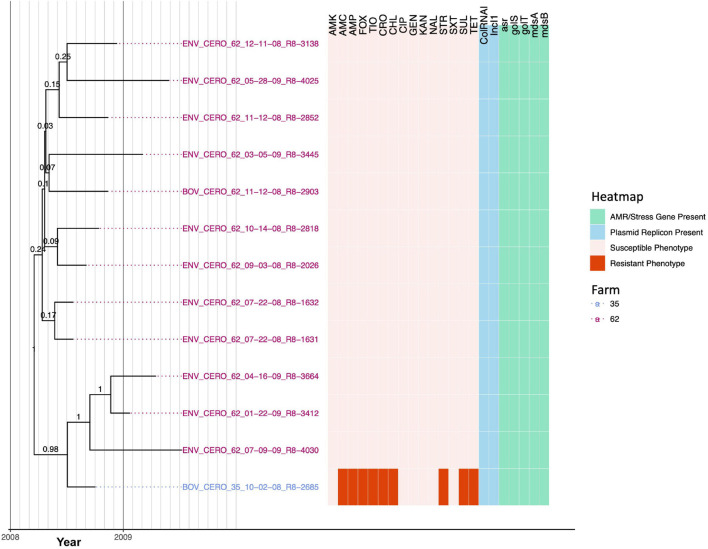
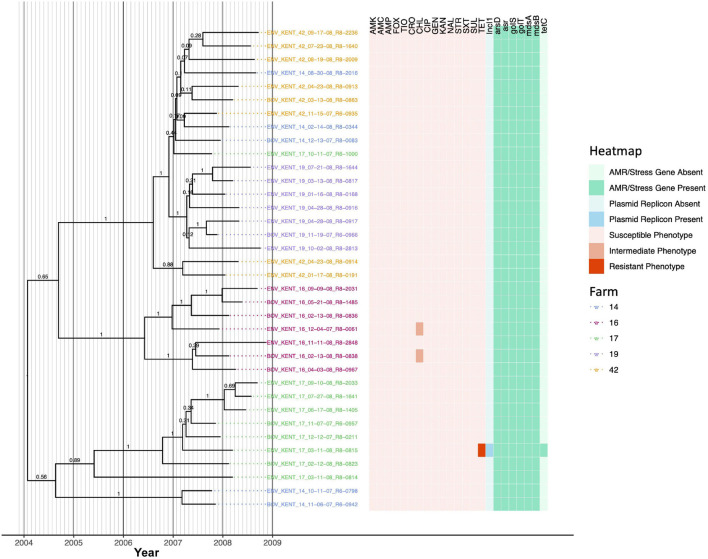
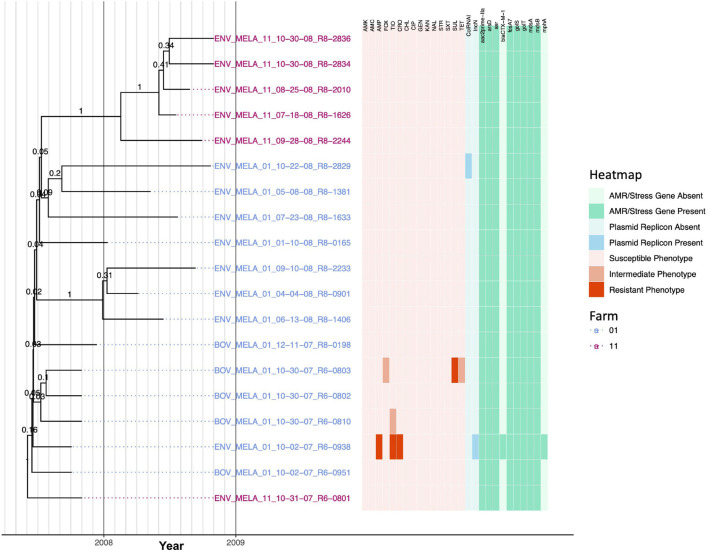
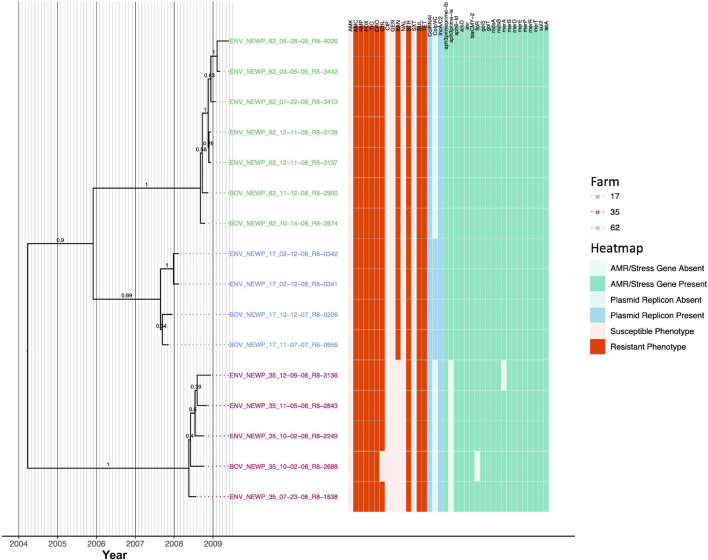
Livestock represent a possible reservoir for facilitating the transmission of the zoonotic foodborne pathogen to humans; there is also concern that strains can acquire resistance to antimicrobials in the farm environment. Here, whole-genome sequencing (WGS) was used to characterize strains ( = 128) isolated from healthy dairy cattle and their associated environments on 13 New York State farms to assess the diversity and microevolution of this important pathogen at the level of the individual herd. Additionally, the accuracy and concordance of multiple tools are assessed, including: (i) two serotyping tools, (ii) combinations of five antimicrobial resistance (AMR) determinant detection tools and one to five AMR determinant databases, and (iii) one antimicrobial minimum inhibitory concentration (MIC) prediction tool. For the isolates sequenced here, serotyping methods outperformed traditional serotyping and resolved all un-typable and/or ambiguous serotype assignments. Serotypes assigned showed greater congruency with the whole-genome phylogeny than traditional serotype assignments, and methods showed high concordance (99% agreement). AMR determinant detection methods additionally showed a high degree of concordance, regardless of the pipeline or database used (≥98% agreement among susceptible/resistant assignments for all pipeline/database combinations). For AMR detection methods that relied exclusively on nucleotide BLAST, accuracy could be maximized by using a range of minimum nucleotide identity and coverage thresholds, with thresholds of 75% nucleotide identity and 50-60% coverage adequate for most pipeline/database combinations. characterization of the microevolution and AMR dynamics of each of six serotype groups ( Anatum, Cerro, Kentucky, Meleagridis, Newport, Typhimurium/Typhimurium variant Copenhagen) revealed that some lineages were strongly associated with individual farms, while others were distributed across multiple farms. Numerous AMR determinant acquisition and loss events were identified, including the recent acquisition of cephalosporin resistance-conferring - and -type beta-lactamases. The results presented here provide high-resolution insight into the temporal dynamics of AMR at the scale of the individual farm and highlight both the strengths and limitations of WGS in tracking zoonotic pathogens and their associated AMR determinants at the livestock-human interface.
家畜可能是促进人畜共患食源性病原体传播给人类的一个潜在宿主;人们还担心菌株可能在农场环境中获得对抗微生物药物的耐药性。在此,利用全基因组测序(WGS)对从纽约州13个农场的健康奶牛及其相关环境中分离出的菌株(n = 128)进行特征分析,以评估这种重要病原体在个体畜群水平上的多样性和微观进化。此外,还评估了多种工具的准确性和一致性,包括:(i)两种血清分型工具,(ii)五种抗菌药物耐药性(AMR)决定簇检测工具与一至五个AMR决定簇数据库的组合,以及(iii)一种抗菌药物最低抑菌浓度(MIC)预测工具。对于此处测序的分离株,WGS血清分型方法优于传统血清分型方法,并解决了所有无法分型和/或模糊的血清型分类问题。所分配的血清型与WGS全基因组系统发育的一致性高于传统血清型分类,并且WGS方法显示出高度一致性(99%的一致性)。AMR决定簇检测方法也显示出高度一致性,无论使用何种流程或数据库(所有流程/数据库组合的敏感/耐药分类之间的一致性≥98%)。对于仅依赖核苷酸BLAST的AMR检测方法,通过使用一系列最低核苷酸同一性和覆盖阈值可使准确性最大化,对于大多数流程/数据库组合而言,75%的核苷酸同一性和50 - 60%的覆盖阈值就足够了。对六个血清型组(阿纳托姆、塞罗、肯塔基、火鸡、纽波特、鼠伤寒/鼠伤寒变种哥本哈根)中每一组的微观进化和AMR动态特征分析表明,一些谱系与个别农场密切相关,而其他谱系则分布在多个农场。鉴定出了许多AMR决定簇的获得和丢失事件,包括最近获得的赋予头孢菌素耐药性的blaCTX-M和blaCMY型β-内酰胺酶。此处呈现的结果提供了在个体农场规模上对AMR沙门氏菌时间动态的高分辨率洞察,并突出了WGS在追踪人畜共患病原体及其在牲畜-人类界面处相关AMR决定簇方面的优势和局限性。