Department of Statistics, Florida State University, 600 W College AVE, Tallahassee, FL 32306, United States.
School of Mathematics, Jilin University, 2699 Qianjin ST, Changchun, Jilin 130012, China.
Bioinformatics. 2023 Jun 1;39(6). doi: 10.1093/bioinformatics/btad366.
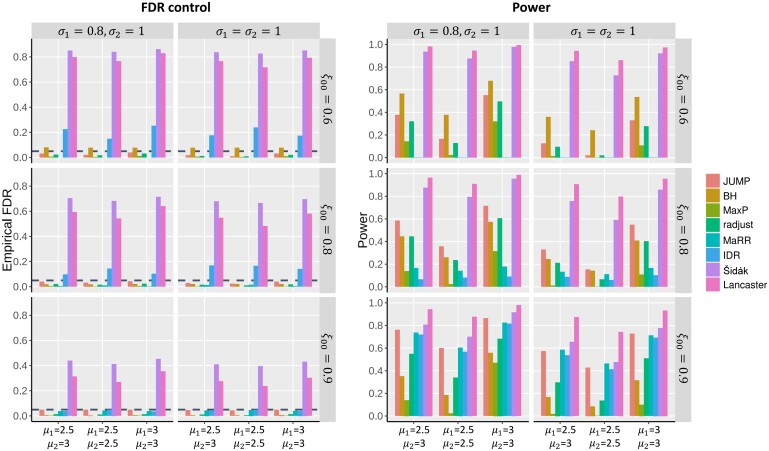
Replicability is the cornerstone of scientific research. The current statistical method for high-dimensional replicability analysis either cannot control the false discovery rate (FDR) or is too conservative.
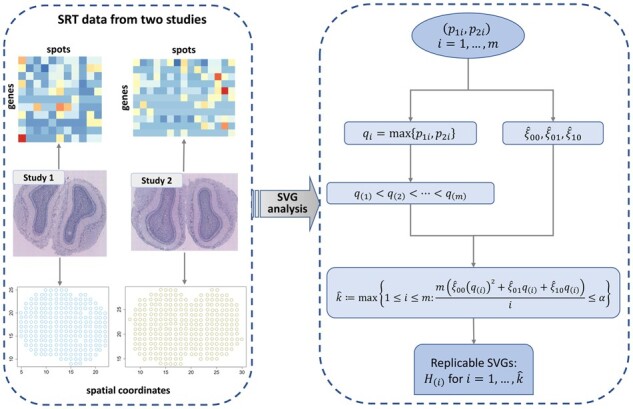
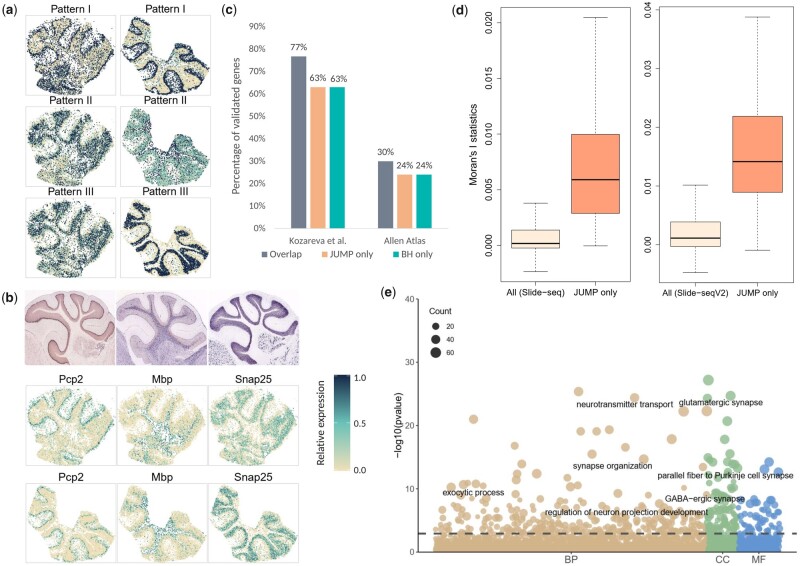
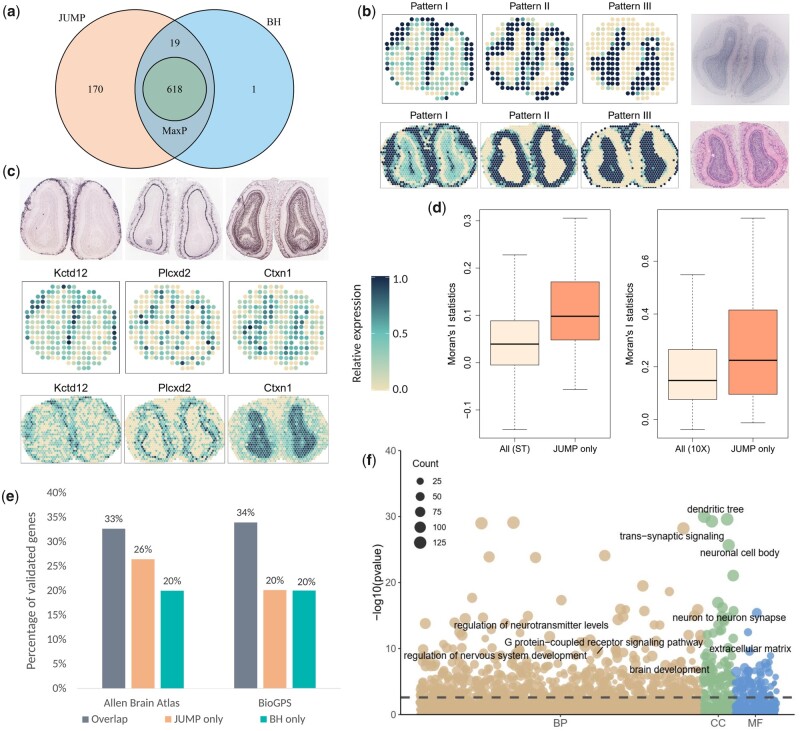
We propose a statistical method, JUMP, for the high-dimensional replicability analysis of two studies. The input is a high-dimensional paired sequence of p-values from two studies and the test statistic is the maximum of p-values of the pair. JUMP uses four states of the p-value pairs to indicate whether they are null or non-null. Conditional on the hidden states, JUMP computes the cumulative distribution function of the maximum of p-values for each state to conservatively approximate the probability of rejection under the composite null of replicability. JUMP estimates unknown parameters and uses a step-up procedure to control FDR. By incorporating different states of composite null, JUMP achieves a substantial power gain over existing methods while controlling the FDR. Analyzing two pairs of spatially resolved transcriptomic datasets, JUMP makes biological discoveries that otherwise cannot be obtained by using existing methods.
An R package JUMP implementing the JUMP method is available on CRAN (https://CRAN.R-project.org/package=JUMP).
可重复性是科学研究的基石。目前用于高维可重复性分析的统计方法要么无法控制错误发现率(FDR),要么过于保守。
我们提出了一种统计方法 JUMP,用于两项研究的高维可重复性分析。输入是两项研究中高维配对的 p 值序列,检验统计量是配对中 p 值的最大值。JUMP 使用 p 值对的四种状态来表示它们是无效还是有效。在隐藏状态的条件下,JUMP 计算每个状态下 p 值最大值的累积分布函数,以保守地近似复合无效性下的拒绝概率。JUMP 估计未知参数并使用逐步程序来控制 FDR。通过合并复合无效性的不同状态,JUMP 在控制 FDR 的同时获得了比现有方法更高的功效。通过分析两对空间分辨转录组数据集,JUMP 做出了其他方法无法获得的生物学发现。
一个实现 JUMP 方法的 R 包 JUMP 可在 CRAN(https://CRAN.R-project.org/package=JUMP)上获得。