Institute of Plant Biology, Biological Research Centre, Szeged, Hungary.
Department of Biotechnology, University of Szeged, Szeged, Hungary.
ISME J. 2023 Aug;17(8):1326-1339. doi: 10.1038/s41396-023-01448-3. Epub 2023 Jun 7.
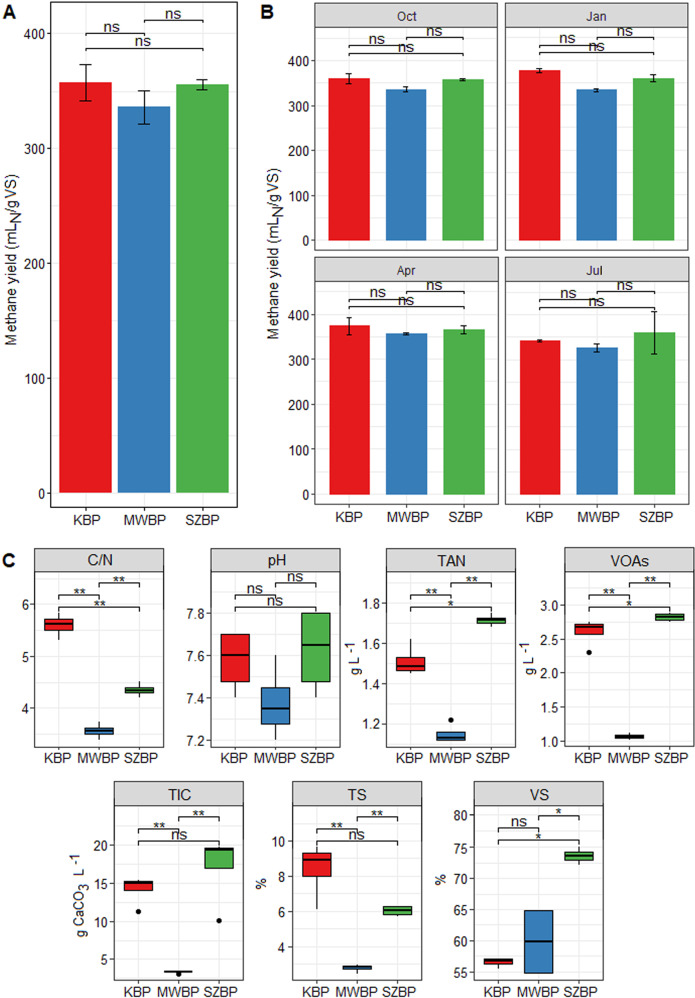
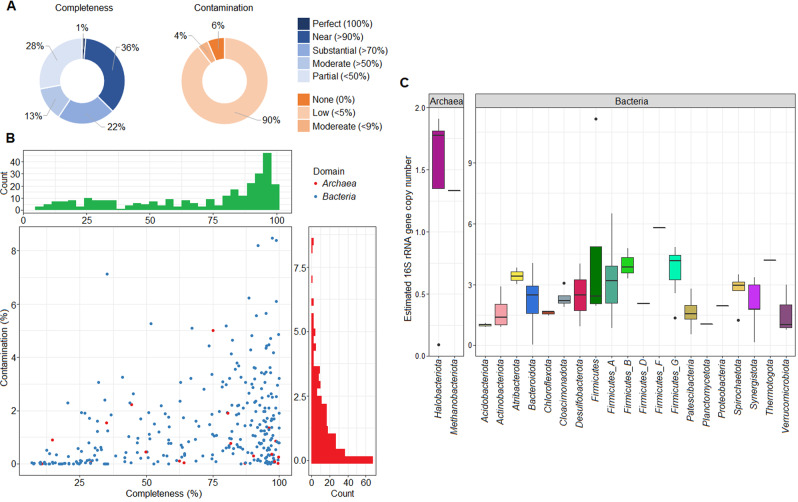
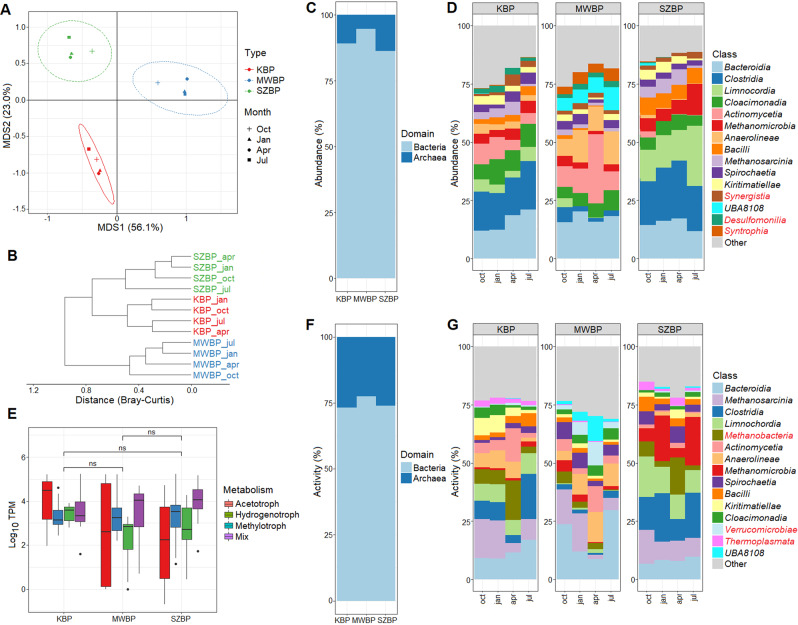
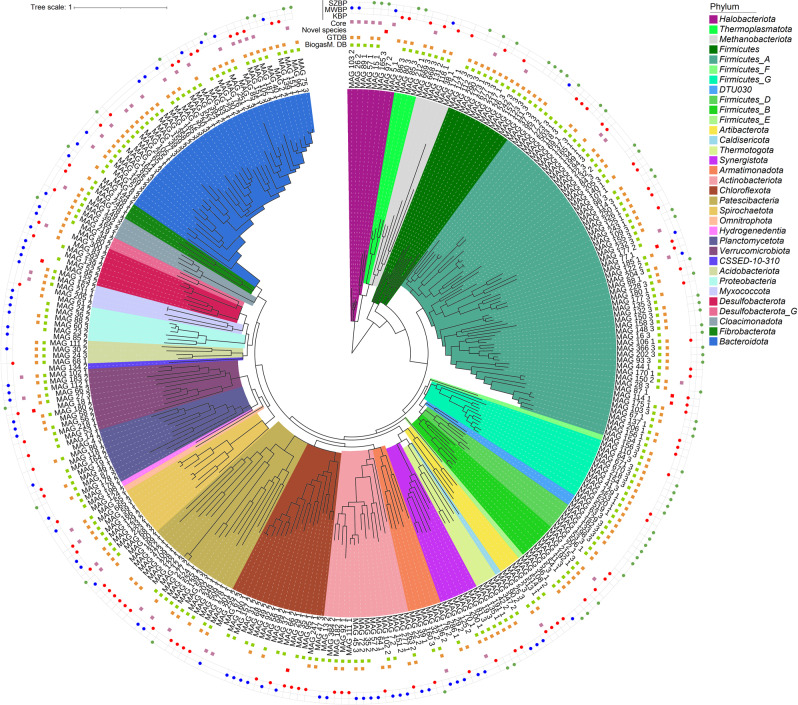
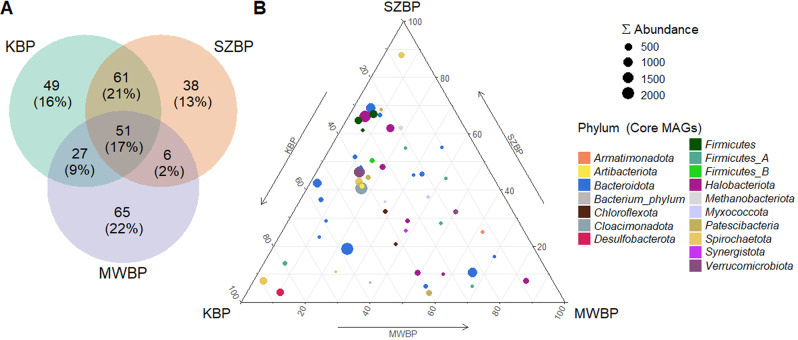
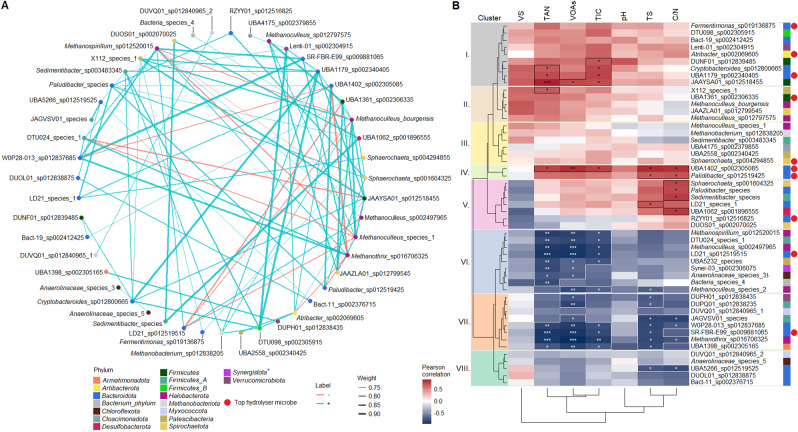
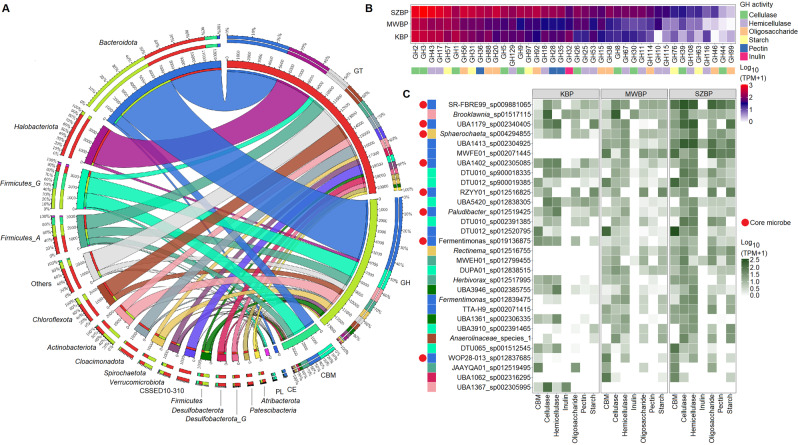
Multi-omics analysis is a powerful tool for the detection and study of inter-kingdom interactions, such as those between bacterial and archaeal members of complex biogas-producing microbial communities. In the present study, the microbiomes of three industrial-scale biogas digesters, each fed with different substrates, were analysed using a machine-learning guided genome-centric metagenomics framework complemented with metatranscriptome data. This data permitted us to elucidate the relationship between abundant core methanogenic communities and their syntrophic bacterial partners. In total, we detected 297 high-quality, non-redundant metagenome-assembled genomes (nrMAGs). Moreover, the assembled 16 S rRNA gene profiles of these nrMAGs showed that the phylum Firmicutes possessed the highest copy number, while the representatives of the archaeal domain had the lowest. Further investigation of the three anaerobic microbial communities showed characteristic alterations over time but remained specific to each industrial-scale biogas plant. The relative abundance of various microorganisms as revealed by metagenome data was independent from corresponding metatranscriptome activity data. Archaea showed considerably higher activity than was expected from their abundance. We detected 51 nrMAGs that were present in all three biogas plant microbiomes with different abundances. The core microbiome correlated with the main chemical fermentation parameters, and no individual parameter emerged as a predominant shaper of community composition. Various interspecies H/electron transfer mechanisms were assigned to hydrogenotrophic methanogens in the biogas plants that ran on agricultural biomass and wastewater. Analysis of metatranscriptome data revealed that methanogenesis pathways were the most active of all main metabolic pathways.
多组学分析是一种强大的工具,可用于检测和研究跨界相互作用,例如复杂沼气产生微生物群落中细菌和古菌成员之间的相互作用。在本研究中,使用机器学习指导的基于基因组的宏基因组学框架和宏转录组数据,分析了三个工业规模沼气消化器的微生物组。这些数据使我们能够阐明丰富的核心产甲烷群落与其协同细菌伙伴之间的关系。总共检测到 297 个高质量、非冗余的宏基因组组装基因组(nrMAG)。此外,这些 nrMAG 的组装 16S rRNA 基因谱表明厚壁菌门拥有最高的拷贝数,而古菌域的代表则拥有最低的拷贝数。对三个厌氧微生物群落的进一步研究表明,随着时间的推移,它们具有特征性的变化,但仍保持每个工业规模沼气厂的特异性。宏基因组数据揭示的各种微生物的相对丰度与相应的宏转录组活性数据无关。古菌的活性明显高于其丰度所预期的水平。我们检测到 51 个 nrMAG 存在于所有三个沼气厂微生物组中,但其丰度不同。核心微生物组与主要的化学发酵参数相关,没有单个参数成为群落组成的主要塑造者。在以农业生物质和废水为原料的沼气厂中,各种种间 H/电子转移机制被分配给产氢甲烷菌。对宏转录组数据的分析表明,产甲烷途径是所有主要代谢途径中最活跃的途径。