Zhou Yuanzhe, Jiang Yangwei, Chen Shi-Jie
Department of Physics and Astronomy, Department of Biochemistry, Institute of Data Sciences and Informatics, University of Missouri, Columbia, MO 65211-7010, USA.
Wiley Interdiscip Rev Comput Mol Sci. 2022 May-Jun;12(3). doi: 10.1002/wcms.1571. Epub 2021 Aug 16.
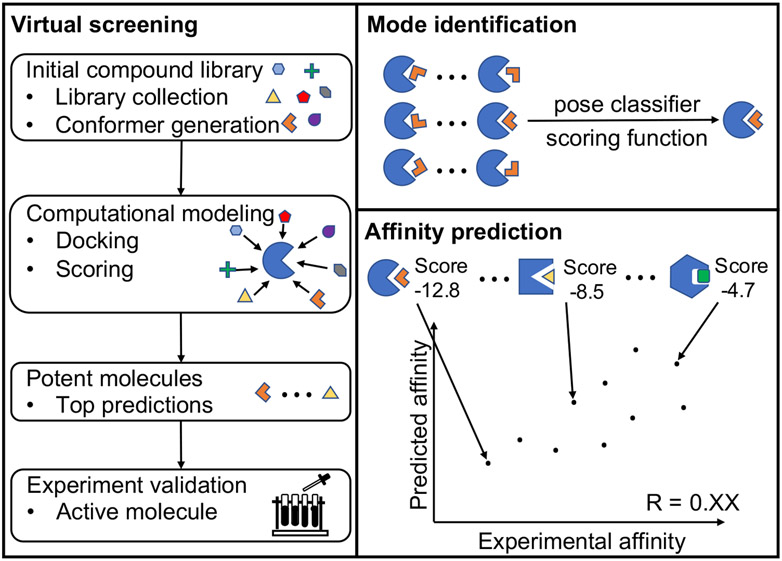
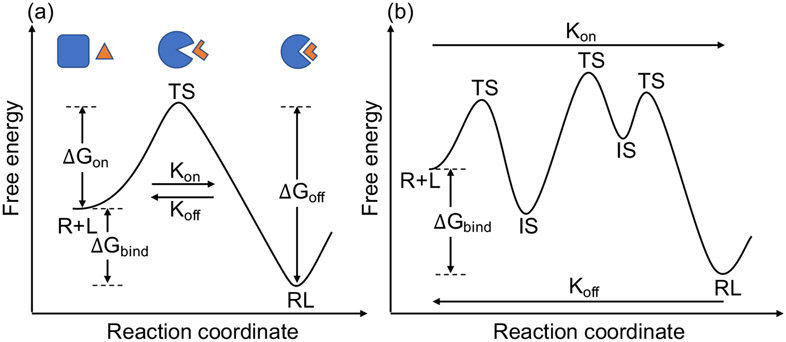
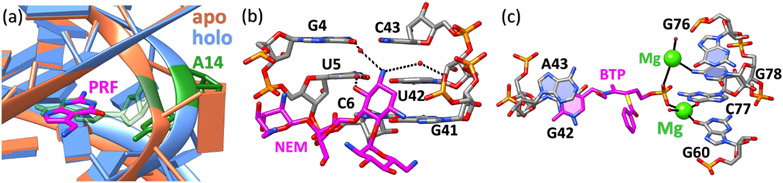
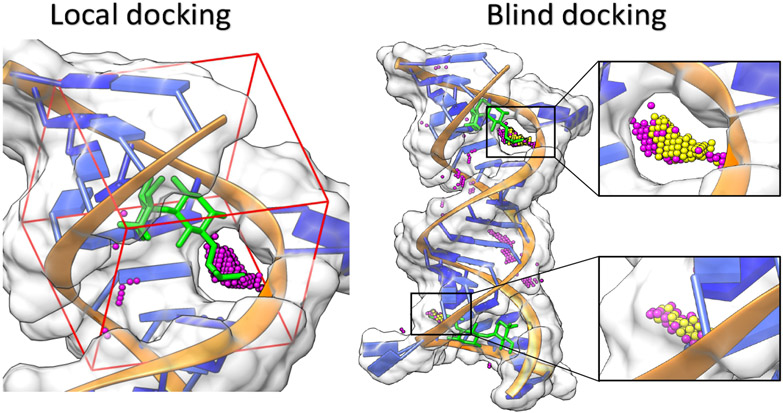
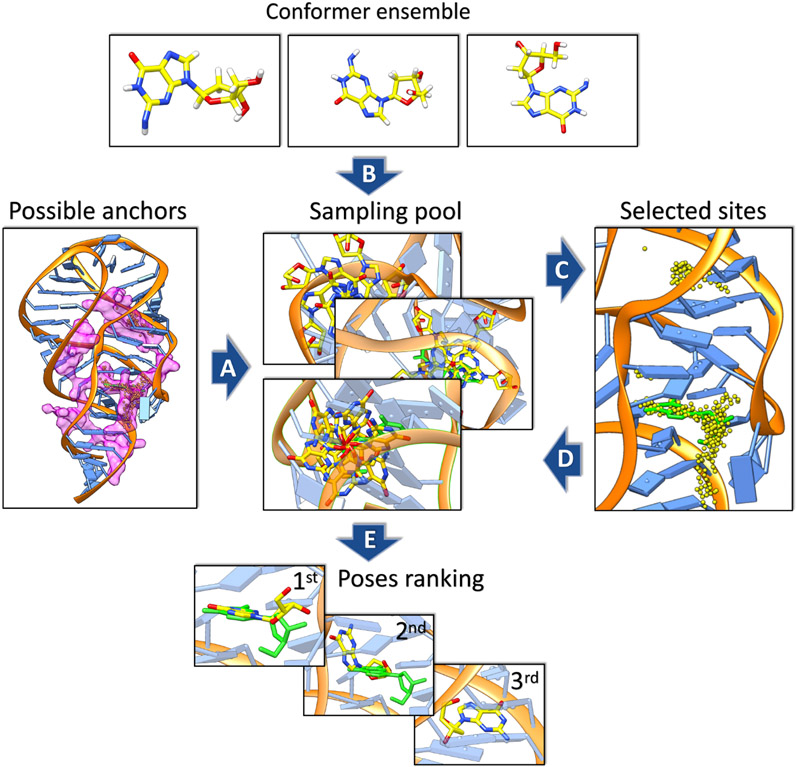
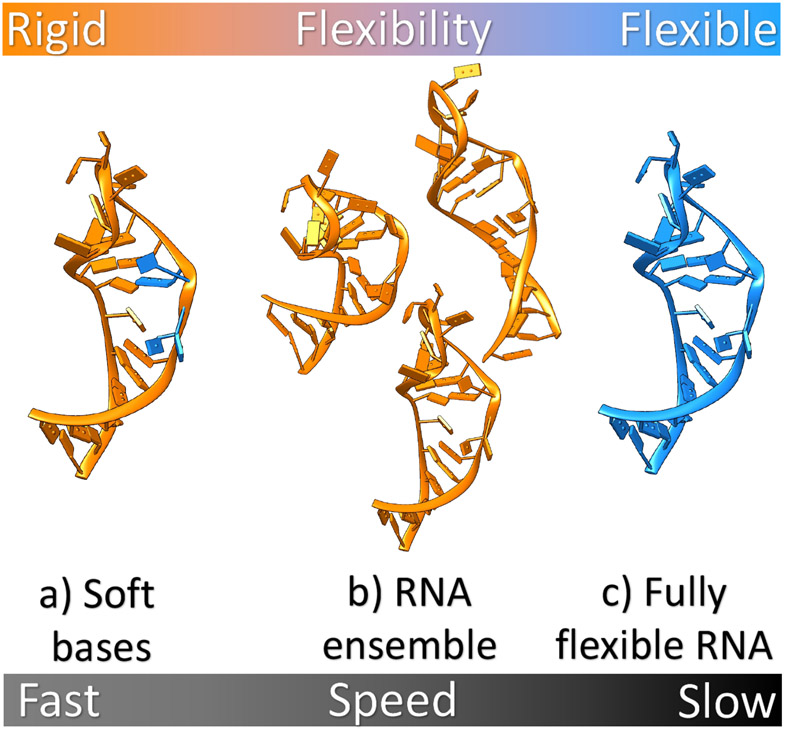
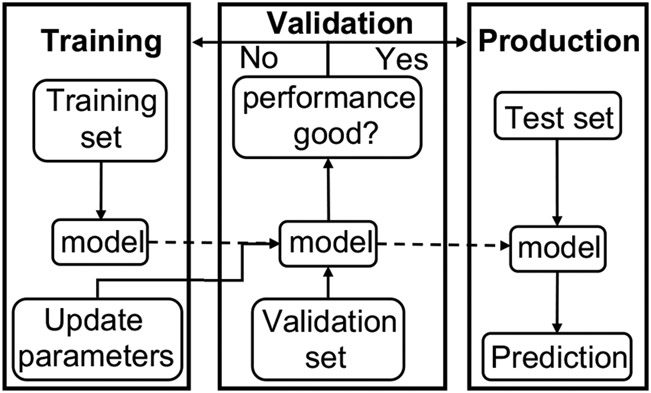
With rapid advances in computer algorithms and hardware, fast and accurate virtual screening has led to a drastic acceleration in selecting potent small molecules as drug candidates. Computational modeling of RNA-small molecule interactions has become an indispensable tool for RNA-targeted drug discovery. The current models for RNA-ligand binding have mainly focused on the docking-and-scoring method. Accurate docking and scoring should tackle four crucial problems: (1) conformational flexibility of ligand, (2) conformational flexibility of RNA, (3) efficient sampling of binding sites and binding poses, and (4) accurate scoring of different binding modes. Moreover, compared with the problem of protein-ligand docking, predicting ligand binding to RNA, a negatively charged polymer, is further complicated by additional effects such as metal ion effects. Thermodynamic models based on physics-based and knowledge-based scoring functions have shown highly encouraging success in predicting ligand binding poses and binding affinities. Recently, kinetic models for ligand binding have further suggested that including dissociation kinetics (residence time) in ligand docking would result in improved performance in estimating drug efficacy. More recently, the rise of deep-learning approaches has led to new tools for predicting RNA-small molecule binding. In this review, we present an overview of the recently developed computational methods for RNA-ligand docking and their advantages and disadvantages.
随着计算机算法和硬件的快速发展,快速准确的虚拟筛选已使筛选有潜力的小分子作为药物候选物的速度大幅加快。RNA与小分子相互作用的计算建模已成为靶向RNA药物研发中不可或缺的工具。当前的RNA-配体结合模型主要集中在对接和评分方法上。准确的对接和评分应解决四个关键问题:(1)配体的构象灵活性,(2)RNA的构象灵活性,(3)结合位点和结合姿势的有效采样,以及(4)不同结合模式的准确评分。此外,与蛋白质-配体对接问题相比,预测配体与带负电荷的聚合物RNA的结合会因金属离子效应等额外效应而更加复杂。基于物理和基于知识的评分函数的热力学模型在预测配体结合姿势和结合亲和力方面已取得了非常令人鼓舞的成功。最近,配体结合的动力学模型进一步表明,在配体对接中纳入解离动力学(停留时间)将在估计药物疗效方面提高性能。最近,深度学习方法的兴起带来了预测RNA-小分子结合的新工具。在这篇综述中,我们概述了最近开发的用于RNA-配体对接的计算方法及其优缺点。