Gedikbasi Asuman, Toksoy Guven, Karaca Meryem, Gulec Cagri, Balci Mehmet Cihan, Gunes Dilek, Gunes Seda, Aslanger Ayca Dilruba, Unverengil Gokcen, Karaman Birsen, Basaran Seher, Demirkol Mubeccel, Gokcay Gulden Fatma, Uyguner Zehra Oya
Department of Pediatric Basic Sciences, Institute of Child Health Istanbul University, Istanbul, Türkiye.
Division of Pediatric Nutrition and Metabolism, Department of Pediatrics, Istanbul Faculty of Medicine, Istanbul University, Istanbul, Türkiye.
Front Genet. 2023 Jun 12;14:1191159. doi: 10.3389/fgene.2023.1191159. eCollection 2023.
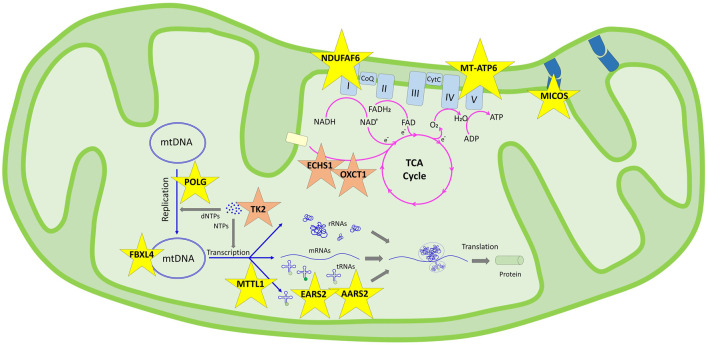
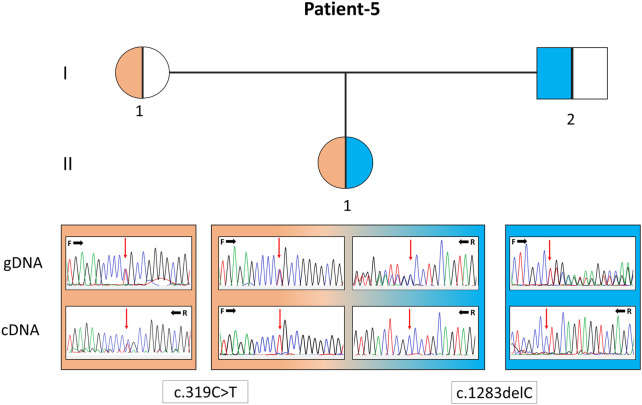
Mitochondrial diseases are the most common group of inherited metabolic disorders, causing difficulties in definite diagnosis due to clinical and genetic heterogeneity. Clinical components are predominantly associated with pathogenic variants shown in nuclear or mitochondrial genomes that affect vital respiratory chain function. The development of high-throughput sequencing technologies has accelerated the elucidation of the genetic etiology of many genetic diseases that previously remained undiagnosed. Thirty affected patients from 24 unrelated families with clinical, radiological, biochemical, and histopathological evaluations considered for mitochondrial diseases were investigated. DNA isolated from the peripheral blood samples of probands was sequenced for nuclear exome and mitochondrial DNA (mtDNA) analyses. MtDNA sequencing was also performed from the muscle biopsy material in one patient. For segregation, Sanger sequencing is performed for pathogenic alterations in five other affected family members and healthy parents. Exome sequencing revealed 14 different pathogenic variants in nine genes encoding mitochondrial function peptides (, and ) in 12 patients from nine families and four variants in genes encoding important for muscle structure ( and ) in six patients from four families. Three probands carried pathogenic mtDNA variations in two genes ( and ). Nine variants in five genes are reported for the first time with disease association: (: c.277C>T/p.(R93*), c.845C>G/p.(S282C); : c.319C>T/p.(R107C), c.1283delC/p.(P428Lfs*); : c.161G>A/p.(R54His); c.202G>A/p.(E68Lys); : c.479delA/p.(N162Ifs*27); and : c.1370C>T/p.(T457I), c.1173-139G>T/p.(?). Bi-genomic DNA sequencing clarified genetic etiology in 67% (16/24) of the families. Diagnostic utility by mtDNA sequencing in 13% (3/24) and exome sequencing in 54% (13/24) of the families prioritized searching for nuclear genome pathologies for the first-tier test. Weakness and muscle wasting observed in 17% (4/24) of the families underlined that limb-girdle muscular dystrophy, similar to mitochondrial myopathy, is an essential point for differential diagnosis. The correct diagnosis is crucial for comprehensive genetic counseling of families. Also, it contributes to making treatment-helpful referrals, such as ensuring early access to medication for patients with mutations in the TK2 gene.
线粒体疾病是最常见的遗传性代谢紊乱疾病组,由于临床和遗传异质性,导致明确诊断困难。临床症状主要与核基因组或线粒体基因组中显示的致病变异有关,这些变异会影响重要的呼吸链功能。高通量测序技术的发展加速了许多以前未被诊断的遗传疾病的遗传病因的阐明。对来自24个无关家庭的30名受影响患者进行了调查,这些患者均经过临床、放射学、生化和组织病理学评估以确定是否患有线粒体疾病。从先证者的外周血样本中分离出DNA,进行核外显子组和线粒体DNA(mtDNA)分析。还对一名患者的肌肉活检材料进行了mtDNA测序。为了进行遗传连锁分析,对另外五名受影响家庭成员和健康父母的致病变异进行了桑格测序。外显子组测序在来自九个家庭的12名患者中发现了九个编码线粒体功能肽(、和)的基因中的14种不同的致病变异,以及在来自四个家庭的六名患者中发现了编码对肌肉结构重要的基因(和)中的四种变异。三名先证者在两个基因(和)中携带致病变异的mtDNA。首次报道了五个基因中的九个变异与疾病相关:(:c.277C>T/p.(R93*),c.845C>G/p.(S282C);:c.319C>T/p.(R107C),c.1283delC/p.(P428Lfs*);:c.161G>A/p.(R54His);c.202G>A/p.(E68Lys);:c.479delA/p.(N162Ifs*27);和:c.1370C>T/p.(T457I),c.1173-139G>T/p.(?)。双基因组DNA测序明确了67%(16/24)家庭的遗传病因。13%(3/24)的家庭通过mtDNA测序进行诊断,54%(13/)的家庭通过外显子组测序进行诊断,这优先考虑了在一级检测中寻找核基因组病变。17%(4/24)的家庭中观察到的虚弱和肌肉萎缩强调,与线粒体肌病类似,肢带型肌营养不良是鉴别诊断的要点。正确的诊断对于家庭的全面遗传咨询至关重要。此外,它有助于进行有助于治疗的转诊,例如确保TK2基因突变患者能够尽早获得药物治疗。