Pritzker School of Molecular Engineering, The University of Chicago, Chicago, Illinois 60637, United States.
Department of Chemistry, University of Chicago, Chicago, Illinois 60637, United States.
J Chem Theory Comput. 2023 Jul 11;19(13):4011-4022. doi: 10.1021/acs.jctc.3c00424. Epub 2023 Jun 28.
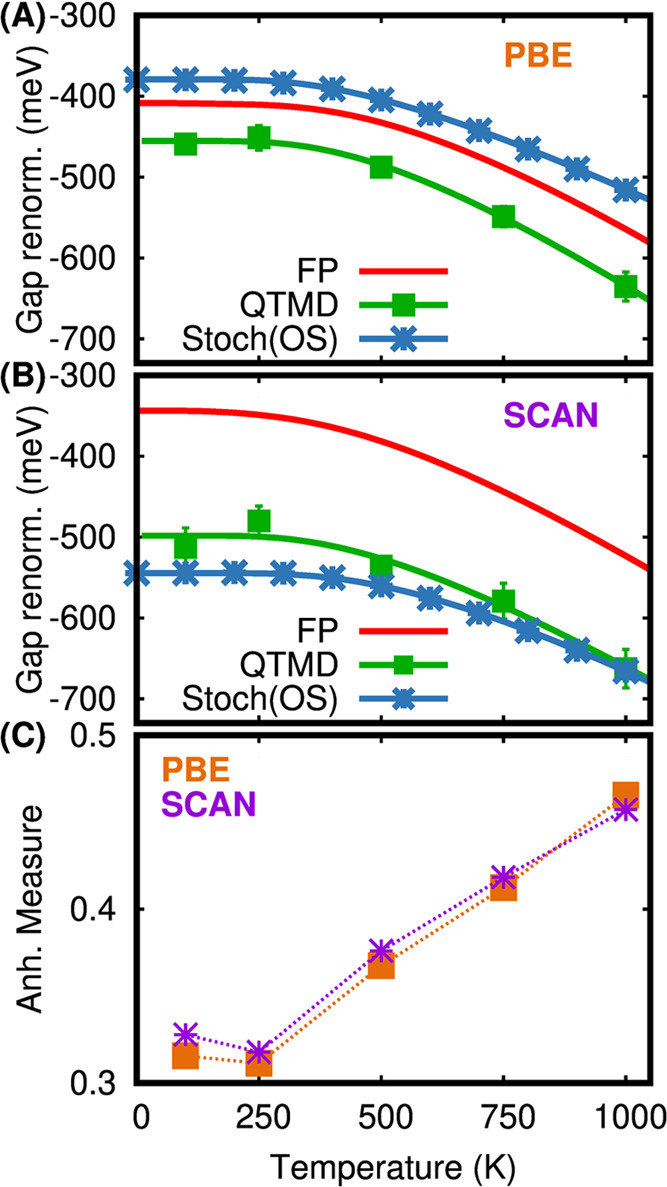
We present a study of molecular crystals, focused on the effect of nuclear quantum motion and anharmonicity on their electronic properties. We consider a system composed of relatively rigid molecules, a diamondoid crystal, and one composed of floppier molecules, NAI-DMAC, a thermally activated delayed fluorescence compound. We compute fundamental electronic gaps at the density functional theory (DFT) level of theory, with the Perdew-Burke-Erzenhof (PBE) and strongly constrained and approximately normed (SCAN) functionals, by coupling first-principles molecular dynamics with a nuclear quantum thermostat. We find a sizable zero-point renormalization (ZPR) of the band gaps, which is much larger in the case of diamondoids (0.6 eV) than for NAI-DMAC (0.22 eV). We show that the frozen phonon (FP) approximation, which neglects intermolecular anharmonic effects, leads to a large error (∼50%) in the calculation of the band gap ZPR. Instead, when using a stochastic method, we obtain results in good agreement with those of our quantum simulations for the diamondoid crystal. However, the agreement is worse for NAI-DMAC where intramolecular anharmonicities contribute to the ZPR. Our results highlight the importance of accurately including nuclear and anharmonic quantum effects to predict the electronic properties of molecular crystals.
我们研究了分子晶体,重点关注核量子运动和非谐性对其电子性质的影响。我们考虑了一个由相对刚性分子组成的系统,即金刚石型晶体,和一个由较柔软分子组成的系统,即 NAI-DMAC,这是一种热活化延迟荧光化合物。我们通过将第一性原理分子动力学与核量子恒温器耦合,在密度泛函理论(DFT)水平上计算了基本的电子能隙,使用了 Perdew-Burke-Erzenhof(PBE)和强约束近似归一化(SCAN)泛函。我们发现带隙的零点能重整化(ZPR)相当大,金刚石型晶体的 ZPR (0.6 eV)比 NAI-DMAC 的 ZPR (0.22 eV)大得多。我们表明,冻结声子(FP)近似忽略了分子间的非谐效应,会导致带隙 ZPR 的计算出现很大的误差(约 50%)。相比之下,当使用随机方法时,我们得到的结果与我们对金刚石型晶体的量子模拟结果非常吻合。然而,对于 NAI-DMAC,分子内非谐性对 ZPR 有贡献,结果的吻合程度较差。我们的结果强调了准确包含核和非谐量子效应以预测分子晶体电子性质的重要性。