Division of Rheumatology, Department of Medicine, University of California, San Francisco, San Francisco, CA, USA.
Biological and Medical Informatics Graduate Program, University of California, San Francisco, San Francisco, CA, USA.
Nat Genet. 2023 Sep;55(9):1512-1522. doi: 10.1038/s41588-023-01465-0. Epub 2023 Aug 10.
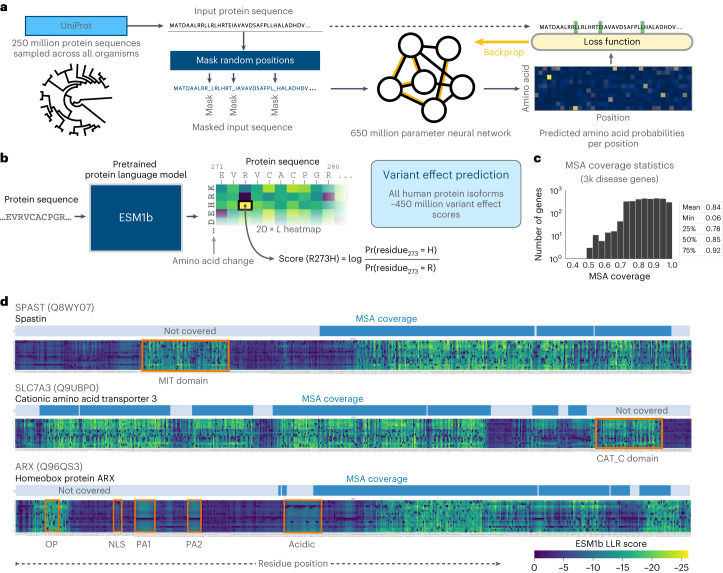
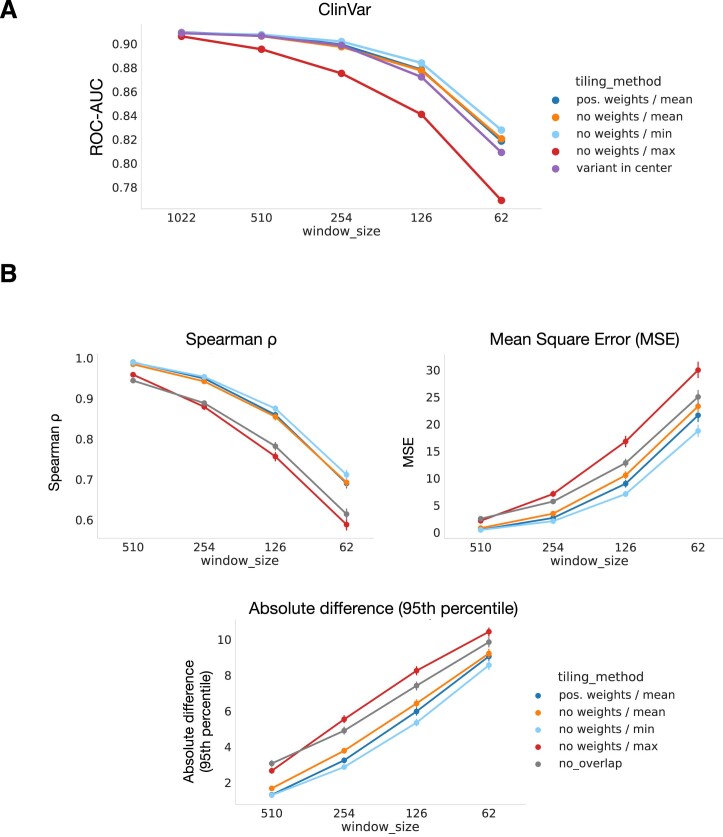
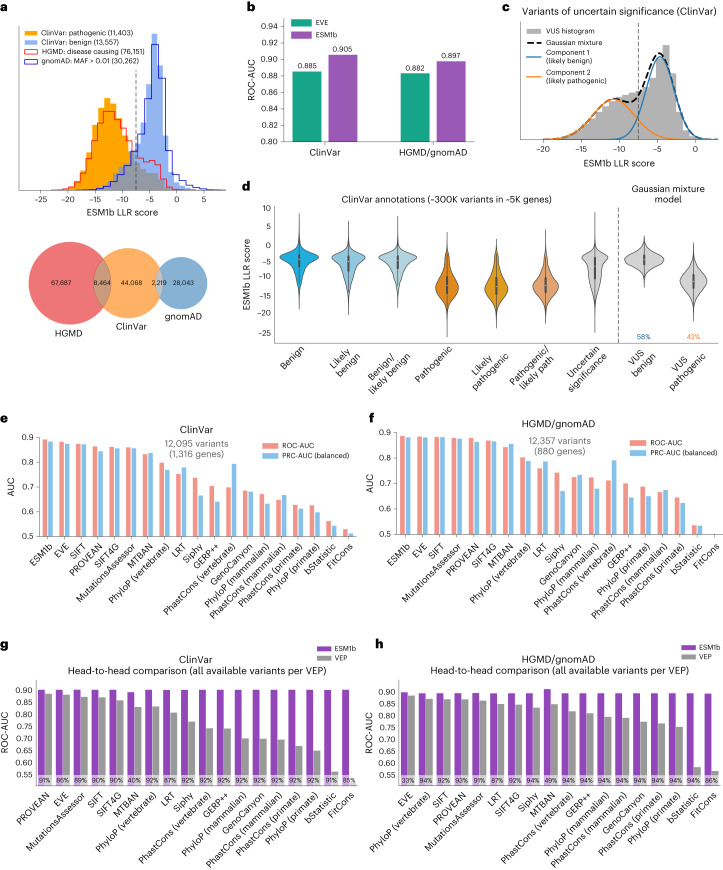
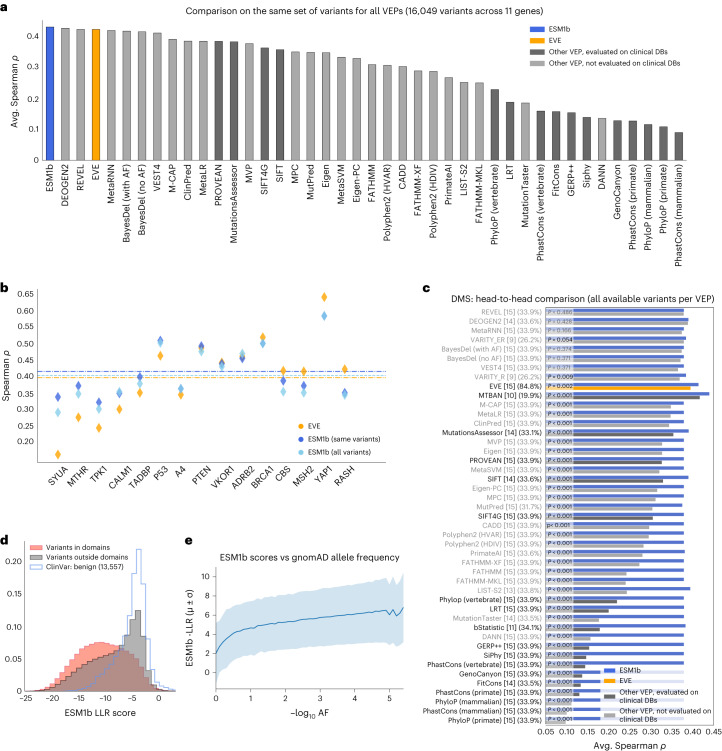
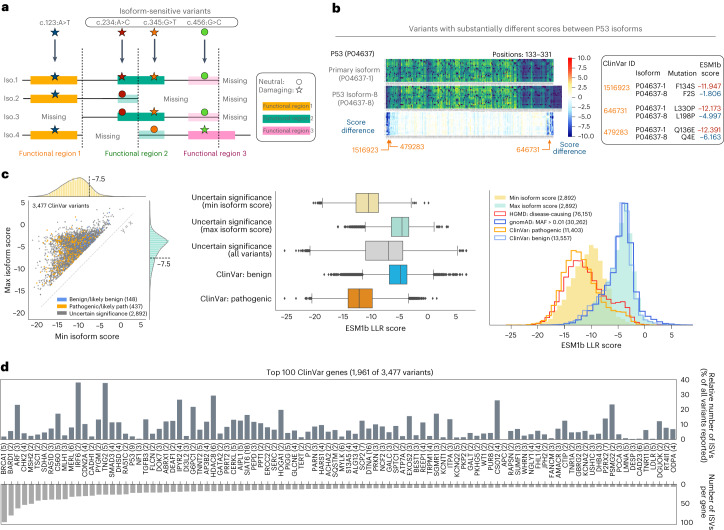
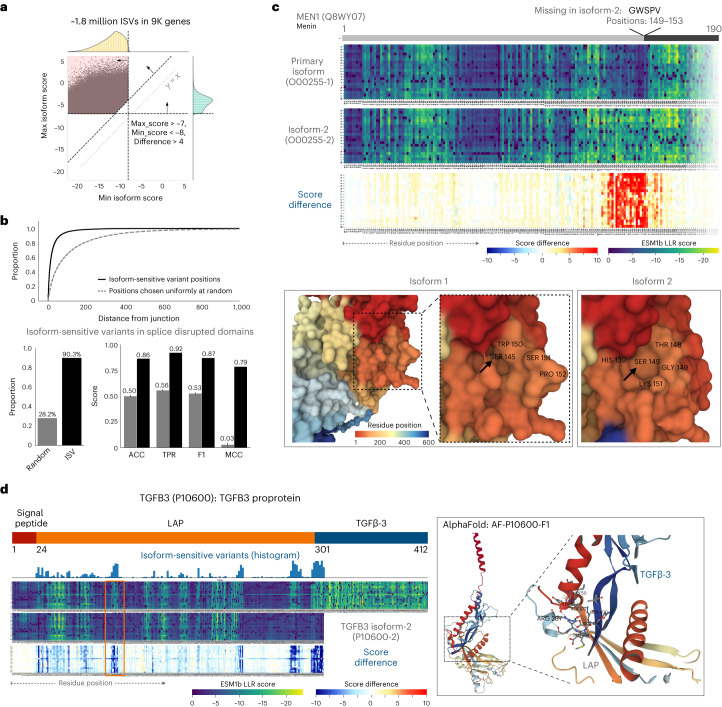
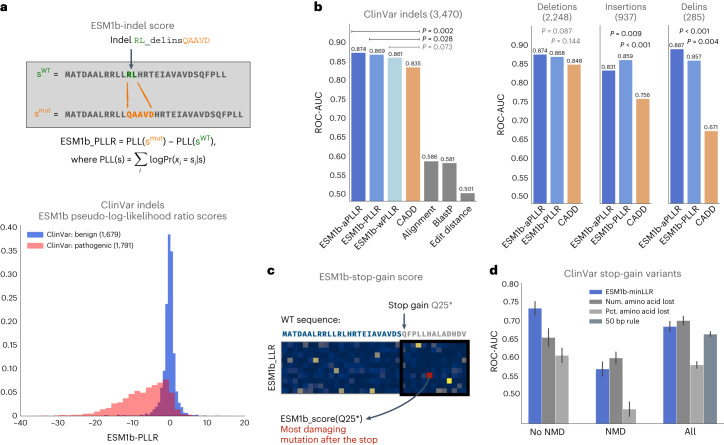
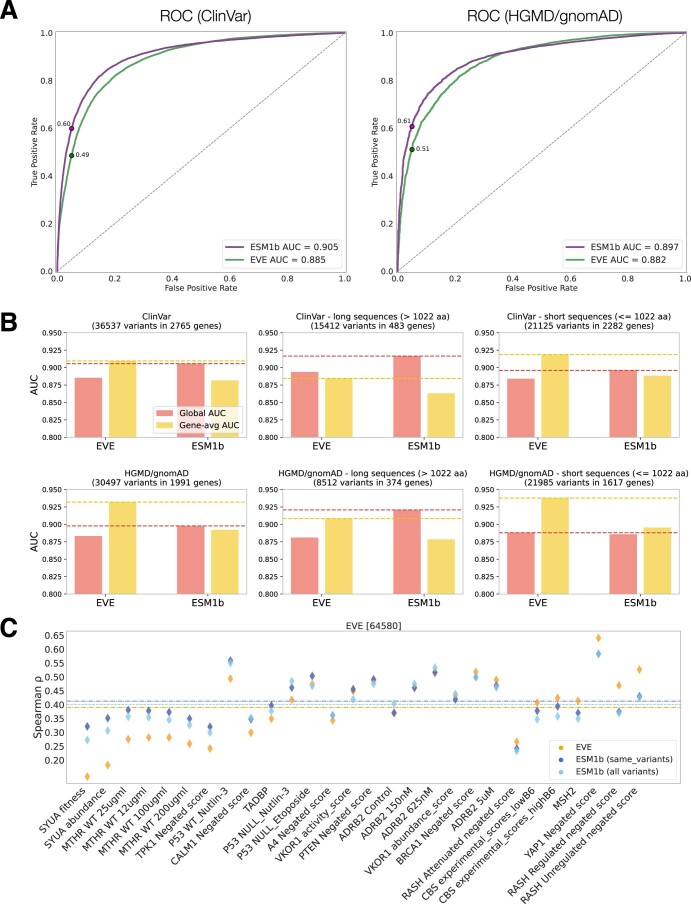
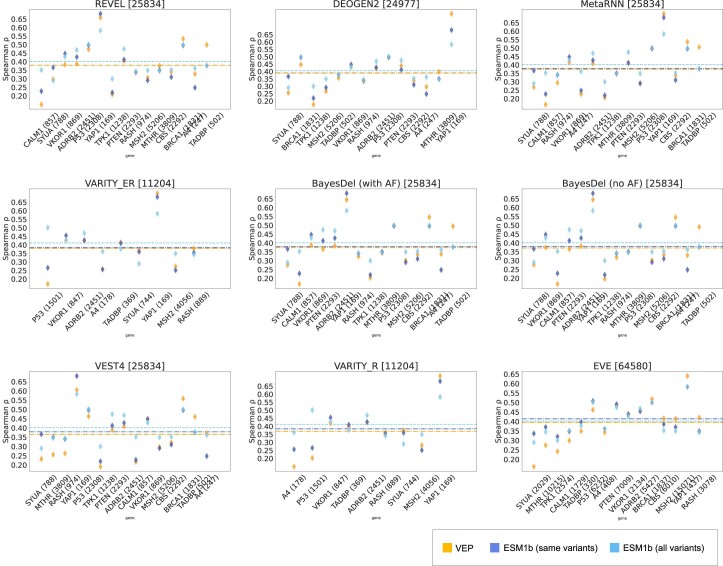
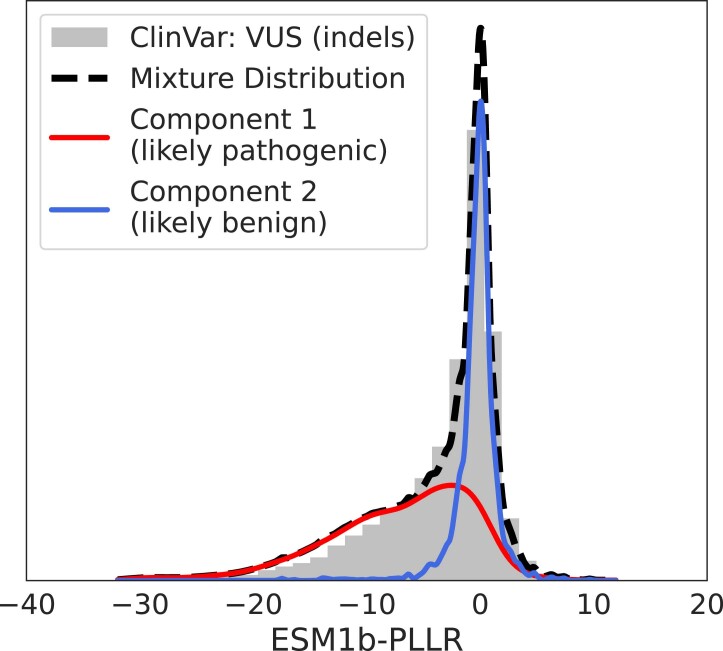
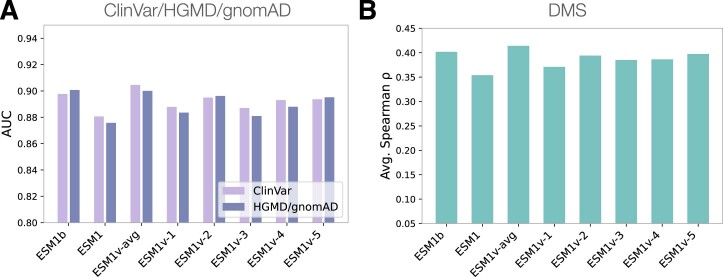
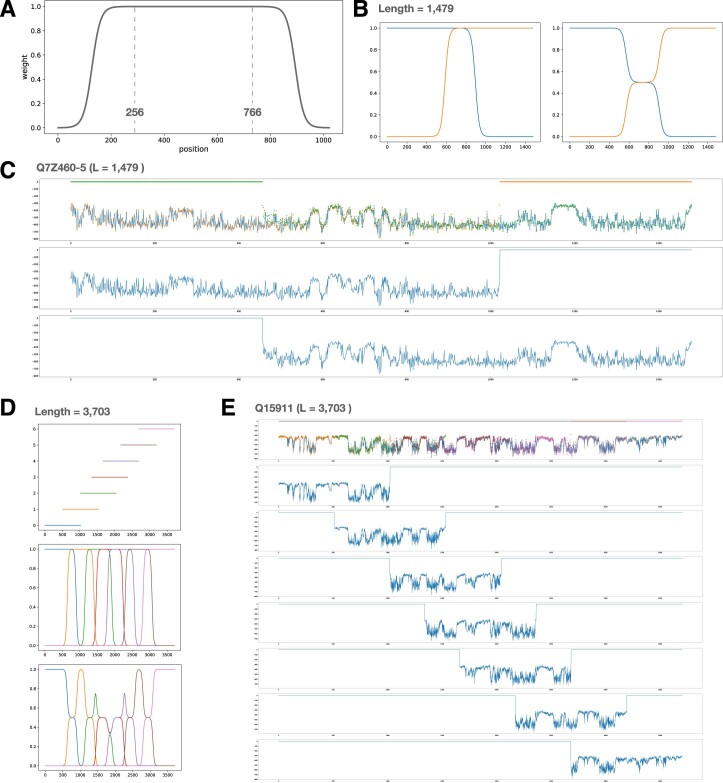
Predicting the effects of coding variants is a major challenge. While recent deep-learning models have improved variant effect prediction accuracy, they cannot analyze all coding variants due to dependency on close homologs or software limitations. Here we developed a workflow using ESM1b, a 650-million-parameter protein language model, to predict all ~450 million possible missense variant effects in the human genome, and made all predictions available on a web portal. ESM1b outperformed existing methods in classifying ~150,000 ClinVar/HGMD missense variants as pathogenic or benign and predicting measurements across 28 deep mutational scan datasets. We further annotated ~2 million variants as damaging only in specific protein isoforms, demonstrating the importance of considering all isoforms when predicting variant effects. Our approach also generalizes to more complex coding variants such as in-frame indels and stop-gains. Together, these results establish protein language models as an effective, accurate and general approach to predicting variant effects.
预测编码变异的影响是一项重大挑战。虽然最近的深度学习模型提高了变异影响预测的准确性,但由于依赖于密切同源物或软件限制,它们无法分析所有编码变异。在这里,我们开发了一个使用 ESM1b 的工作流程,ESM1b 是一个 6.5 亿参数的蛋白质语言模型,用于预测人类基因组中所有约 4.5 亿种可能的错义变异的影响,并在一个网络门户上提供了所有的预测结果。ESM1b 在对约 150,000 个 ClinVar/HGMD 错义变异进行致病性或良性分类以及预测 28 个深度突变扫描数据集的测量方面表现优于现有方法。我们进一步将约 200 万个变体注释为仅在特定蛋白质亚型中具有破坏性,这表明在预测变体影响时考虑所有亚型的重要性。我们的方法也可推广到更复杂的编码变异,如框内缺失和终止增益。总之,这些结果确立了蛋白质语言模型作为预测变异影响的有效、准确和通用的方法。