Institute of Human Genetics, Bonn School of Medicine, University Hospital of Bonn, University of Bonn, Bonn, Germany.
Berlin Institute of Health at Charité-Universitätsmedizin Berlin, Berlin, Germany.
Bioinformatics. 2023 May 4;39(5). doi: 10.1093/bioinformatics/btad280.
Missense variants are a frequent class of variation within the coding genome, and some of them cause Mendelian diseases. Despite advances in computational prediction, classifying missense variants into pathogenic or benign remains a major challenge in the context of personalized medicine. Recently, the structure of the human proteome was derived with unprecedented accuracy using the artificial intelligence system AlphaFold2. This raises the question of whether AlphaFold2 wild-type structures can improve the accuracy of computational pathogenicity prediction for missense variants.
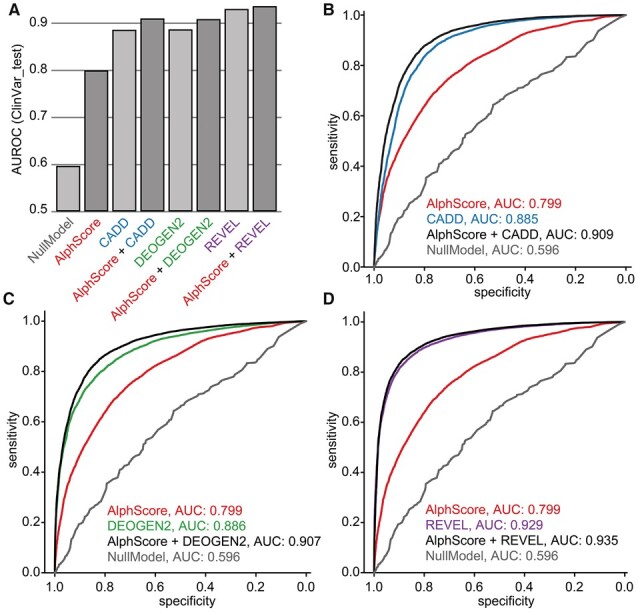
To address this, we first engineered a set of features for each amino acid from these structures. We then trained a random forest to distinguish between relatively common (proxy-benign) and singleton (proxy-pathogenic) missense variants from gnomAD v3.1. This yielded a novel AlphaFold2-based pathogenicity prediction score, termed AlphScore. Important feature classes used by AlphScore are solvent accessibility, amino acid network related features, features describing the physicochemical environment, and AlphaFold2's quality parameter (predicted local distance difference test). AlphScore alone showed lower performance than existing in silico scores used for missense prediction, such as CADD or REVEL. However, when AlphScore was added to those scores, the performance increased, as measured by the approximation of deep mutational scan data, as well as the prediction of expert-curated missense variants from the ClinVar database. Overall, our data indicate that the integration of AlphaFold2-predicted structures can improve pathogenicity prediction of missense variants.
AlphScore, combinations of AlphScore with existing scores, as well as variants used for training and testing are publicly available.
错义变异是编码基因组中常见的变异类型之一,其中一些会导致孟德尔疾病。尽管在计算预测方面取得了进展,但在个性化医疗的背景下,将错义变异分类为致病性或良性仍然是一个主要挑战。最近,使用人工智能系统 AlphaFold2 以前所未有的精度推导出了人类蛋白质组的结构。这就提出了一个问题,即 AlphaFold2 的野生型结构是否可以提高计算错义变异致病性预测的准确性。
为了解决这个问题,我们首先从这些结构中为每个氨基酸设计了一组特征。然后,我们使用随机森林来区分 gnomAD v3.1 中的相对常见(代理良性)和单峰(代理致病性)错义变异。这产生了一种新的基于 AlphaFold2 的致病性预测评分,称为 AlphScore。AlphScore 使用的重要特征类别包括溶剂可及性、氨基酸网络相关特征、描述物理化学环境的特征以及 AlphaFold2 的质量参数(预测局部距离差异测试)。AlphScore 本身的性能低于用于错义预测的现有计算评分,例如 CADD 或 REVEL。然而,当 AlphScore 与这些评分结合使用时,性能会提高,这可以通过对深度突变扫描数据的逼近以及对 ClinVar 数据库中专家 curated 的错义变异的预测来衡量。总体而言,我们的数据表明,整合 AlphaFold2 预测的结构可以提高错义变异的致病性预测。
AlphScore、AlphScore 与现有评分的组合以及用于训练和测试的变体均可供公开使用。