CAS Key Laboratory of Regenerative Biology, Guangdong Provincial Key Laboratory of Stem Cell and Regenerative Medicine, GIBH-HKU Guangdong-Hong Kong Stem Cell and Regenerative Medicine Research Centre, Hong Kong Institute of Science & Innovation, Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, Guangzhou, Guangdong 510530, China.
University of Chinese Academy of Sciences, Beijing 100049, China.
Bioinformatics. 2023 Aug 1;39(8). doi: 10.1093/bioinformatics/btad526.
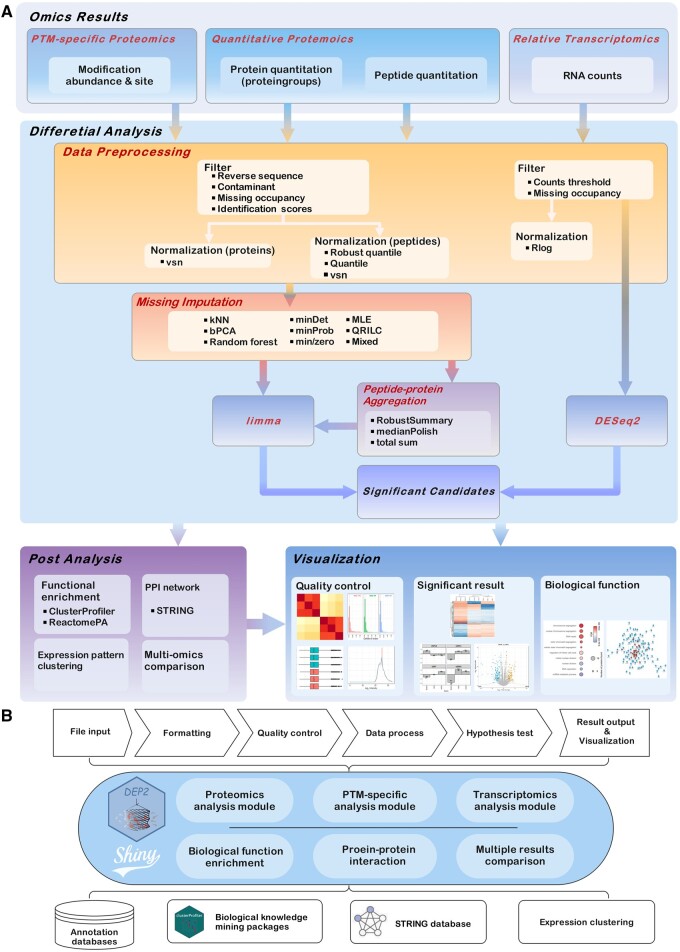
Mass spectrometry (MS)-based proteomics has become the most powerful approach to study the proteome of given biological and clinical samples. Advancements in sample preparation and MS detection have extended the application of proteomics but have also brought new demands on data analysis. Appropriate proteomics data analysis workflow mainly requires quality control, hypothesis testing, functional mining, and visualization. Although there are numerous tools for each process, an efficient and universal tandem analysis toolkit to obtain a quick overall view of various proteomics data is still urgently needed. Here, we present DEP2, an updated version of DEP we previously established, for proteomics data analysis. We amended the analysis workflow by incorporating alternative approaches to accommodate diverse proteomics data, introducing peptide-protein summarization and coupling biological function exploration. In summary, DEP2 is a well-rounded toolkit designed for protein- and peptide-level quantitative proteomics data. It features a more flexible differential analysis workflow and includes a user-friendly Shiny application to facilitate data analysis.
DEP2 is available at https://github.com/mildpiggy/DEP2, released under the MIT license. For further information and usage details, please refer to the package website at https://mildpiggy.github.io/DEP2/.
基于质谱(MS)的蛋白质组学已成为研究特定生物和临床样本蛋白质组的最有力方法。样品制备和 MS 检测的进步扩展了蛋白质组学的应用范围,但也对数据分析提出了新的要求。适当的蛋白质组学数据分析工作流程主要需要质量控制、假设检验、功能挖掘和可视化。尽管每个过程都有许多工具,但仍然迫切需要一个高效和通用的串联分析工具包来快速获得各种蛋白质组学数据的整体视图。在这里,我们展示了 DEP2,这是我们之前建立的 DEP 的更新版本,用于蛋白质组学数据分析。我们通过纳入替代方法来修改分析工作流程,以适应各种蛋白质组学数据,引入肽 - 蛋白质总结并结合生物功能探索。总之,DEP2 是一个全面的工具包,专为蛋白质和肽水平的定量蛋白质组学数据设计。它具有更灵活的差异分析工作流程,并包括一个用户友好的 Shiny 应用程序,以方便数据分析。
DEP2 可在 https://github.com/mildpiggy/DEP2 上获得,根据 MIT 许可证发布。有关更多信息和使用详细信息,请参阅位于 https://mildpiggy.github.io/DEP2/ 的软件包网站。