Zwyer Michaela, Çavusoglu Cengiz, Ghielmetti Giovanni, Pacciarini Maria Lodovica, Scaltriti Erika, Van Soolingen Dick, Dötsch Anna, Reinhard Miriam, Gagneux Sebastien, Brites Daniela
University of Basel, Basel, Switzerland.
Swiss Tropical and Public Health Institute, Basel, Switzerland.
Open Res Eur. 2021 Dec 1;1:100. doi: 10.12688/openreseurope.14029.2. eCollection 2021.
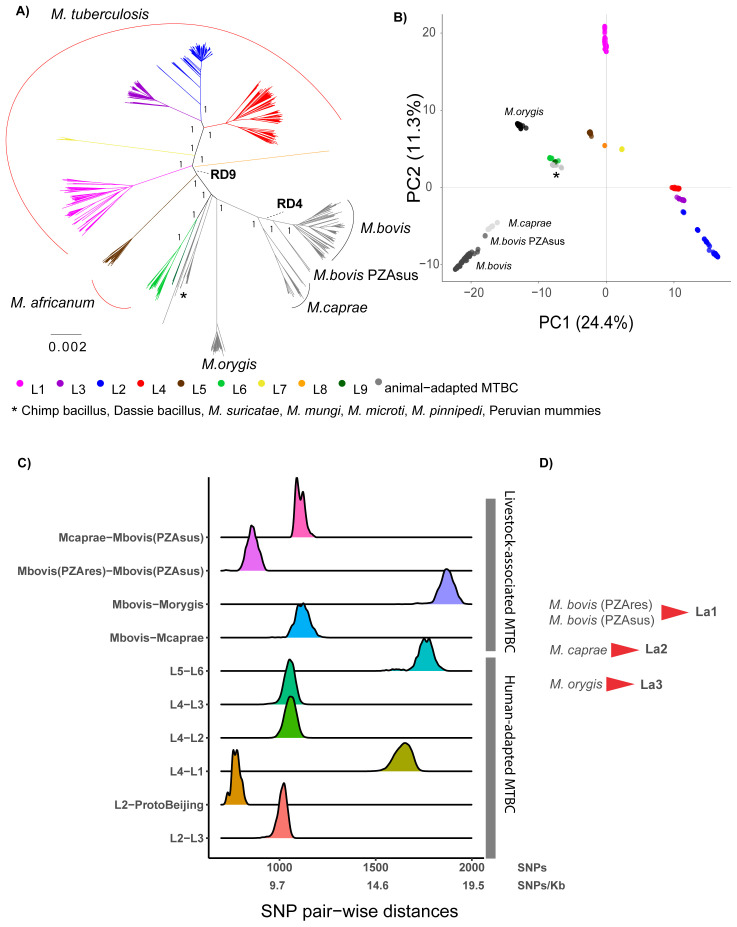
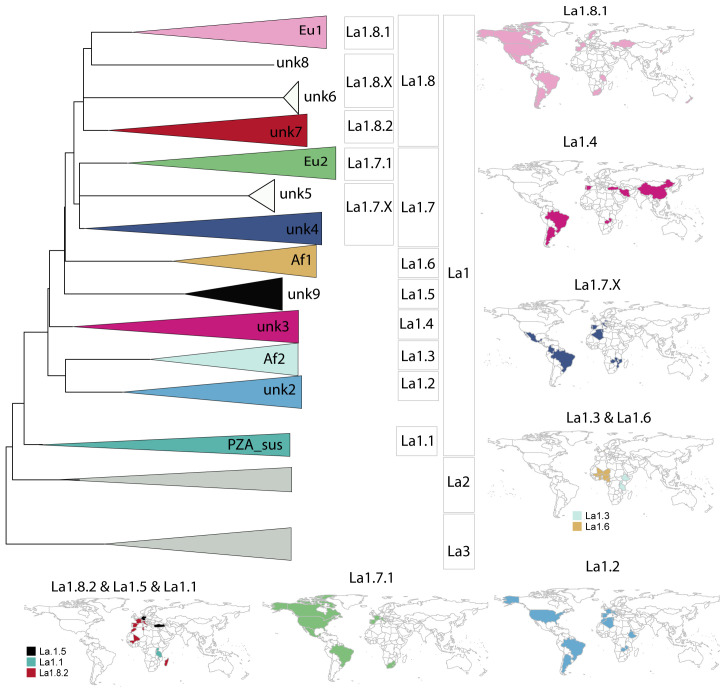
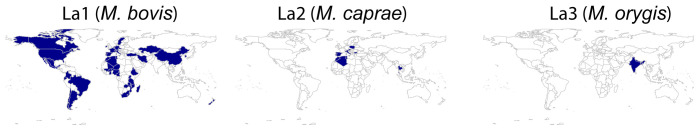
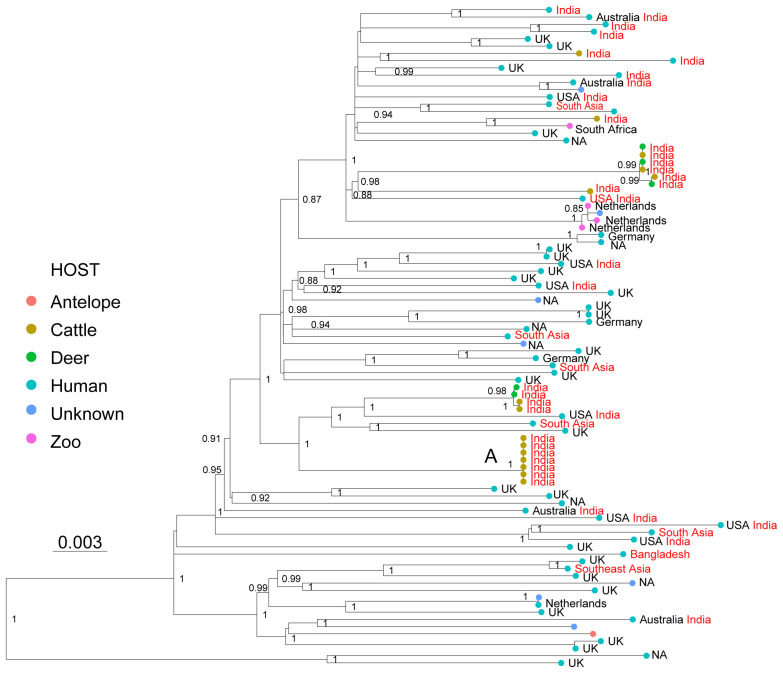
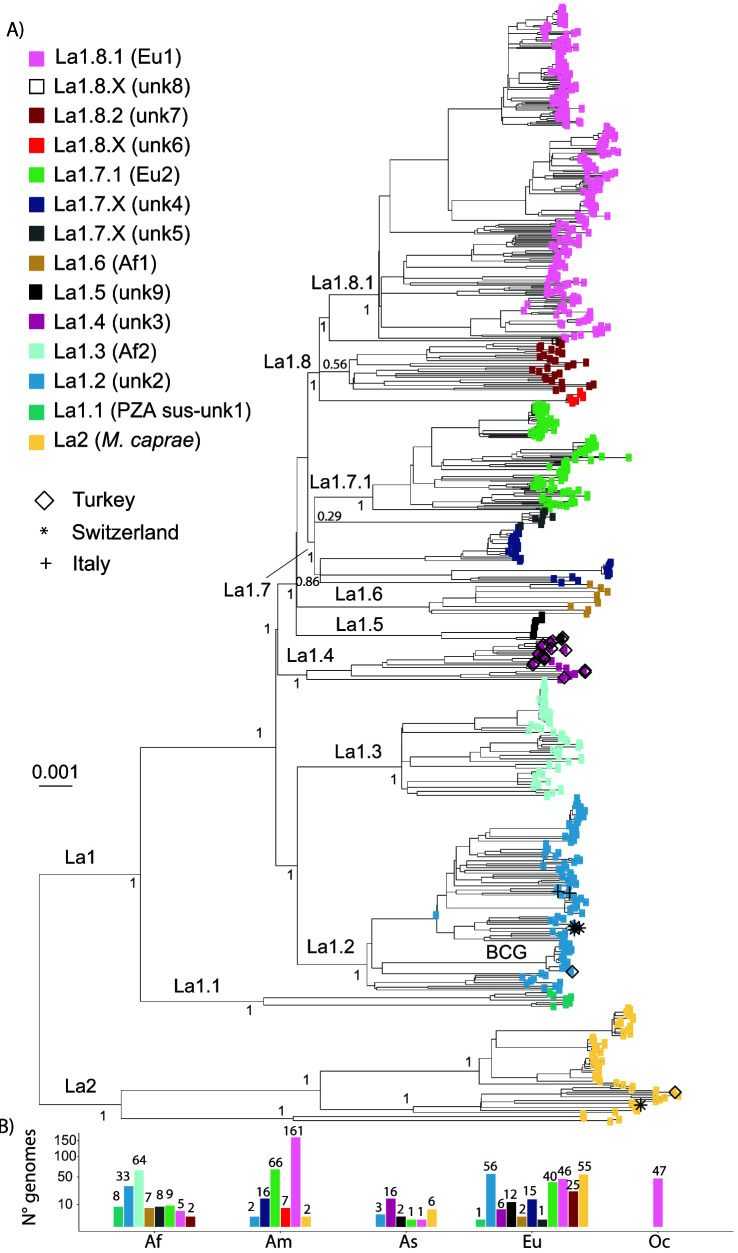
: The bacteria that compose the complex (MTBC) cause tuberculosis (TB) in humans and in different animals, including livestock. Much progress has been made in understanding the population structure of the human-adapted members of the MTBC by combining phylogenetics with genomics. Accompanying the discovery of new genetic diversity, a body of operational nomenclature has evolved to assist comparative and molecular epidemiological studies of human TB. By contrast, for the livestock-associated MTBC members, , and , there has been a lack of comprehensive nomenclature to accommodate new genetic diversity uncovered by emerging phylogenomic studies. We propose to fill this gap by putting forward a new nomenclature covering the main phylogenetic groups within , and . : We gathered a total of 8,736 whole-genome sequences (WGS) from public sources and 39 newly sequenced strains, and selected a subset of 829 WGS, representative of the worldwide diversity of , and . We used phylogenetics and genetic diversity patterns inferred from WGS to define groups. : We propose to divide , and in three main phylogenetic lineages, which we named La1, La2 and La3, respectively. Within La1, we identified several monophyletic groups, which we propose to classify into eight sublineages (La1.1-La1.8). These sublineages differed in geographic distribution, with some being geographically restricted and others globally widespread, suggesting different expansion abilities. To ease molecular characterization of these MTBC groups by the community, we provide phylogenetically informed, single nucleotide polymorphisms that can be used as barcodes for genotyping. These markers were implemented in KvarQ and TB-Profiler, which are platform-independent, open-source tools. : Our results contribute to an improved classification of the genetic diversity within the livestock-associated MTBC, which will benefit future molecular epidemiological and evolutionary studies.
构成复合菌群(MTBC)的细菌可在人类及包括家畜在内的不同动物中引发结核病(TB)。通过将系统发育学与基因组学相结合,在理解MTBC中适应人类的菌群的种群结构方面已取得了很大进展。随着新遗传多样性的发现,一套实用的命名法逐渐发展起来,以协助人类结核病的比较和分子流行病学研究。相比之下,对于与家畜相关的MTBC菌群成员,即牛分枝杆菌、非洲分枝杆菌和田鼠分枝杆菌,缺乏全面的命名法来适应新兴系统基因组学研究所发现的新遗传多样性。我们建议通过提出一种涵盖牛分枝杆菌、非洲分枝杆菌和田鼠分枝杆菌内主要系统发育组的新命名法来填补这一空白。我们从公共来源收集了总共8736个全基因组序列(WGS)和39个新测序菌株,并选择了829个WGS的子集,代表牛分枝杆菌、非洲分枝杆菌和田鼠分枝杆菌的全球多样性。我们利用从WGS推断出的系统发育学和遗传多样性模式来定义菌群。我们建议将牛分枝杆菌、非洲分枝杆菌和田鼠分枝杆菌分为三个主要系统发育谱系,我们分别将其命名为La1、La2和La3。在La1内,我们确定了几个单系群,我们建议将其分类为八个亚谱系(La1.1-La1.8)。这些亚谱系在地理分布上有所不同,有些在地理上受到限制,而有些则广泛分布于全球,这表明它们具有不同的扩张能力。为便于该领域对这些MTBC菌群进行分子特征分析,我们提供了基于系统发育的单核苷酸多态性,可将其用作基因分型的条形码。这些标记已在KvarQ和TB-Profiler中实现,这两个都是独立于平台的开源工具。我们的结果有助于改进对与家畜相关的MTBC内遗传多样性的分类,这将有利于未来的分子流行病学和进化研究。