Li Kaitao, Cardenas-Lizana Paul, Kellner Anna V, Yuan Zhou, Ahn Eunseon, Lyu Jintian, Li Zhenhai, Salaita Khalid, Ahmed Rafi, Zhu Cheng
Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology, Atlanta, Georgia 30332, USA.
Parker H. Petit Institute for Bioengineering and Biosciences, Georgia Institute of Technology, Atlanta, Georgia 30332, USA.
bioRxiv. 2023 Aug 15:2023.08.13.553152. doi: 10.1101/2023.08.13.553152.
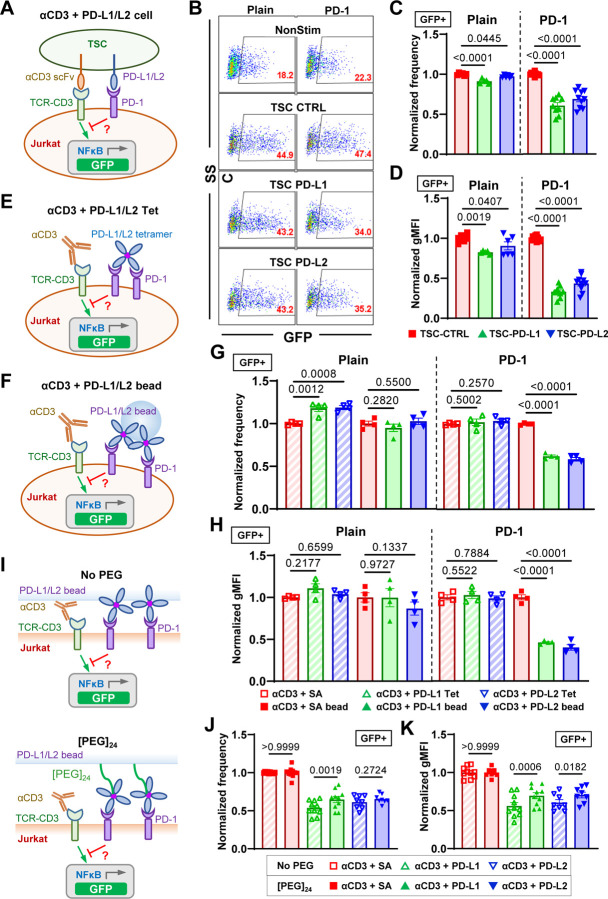
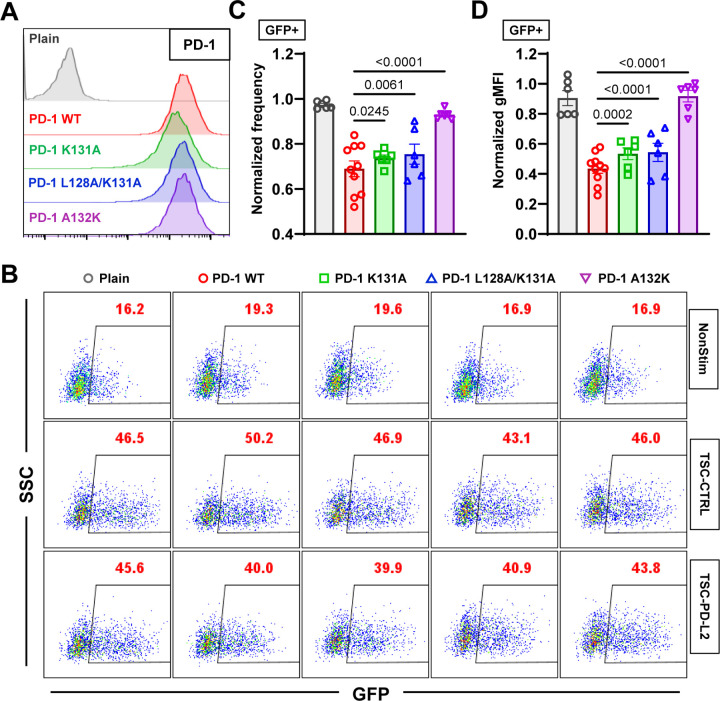
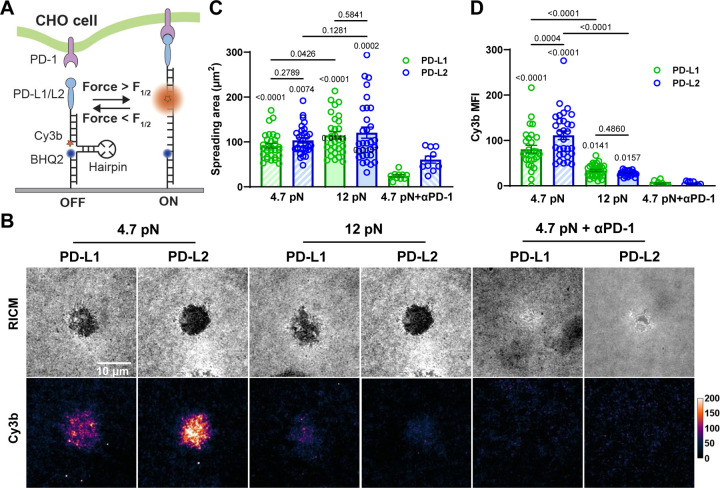
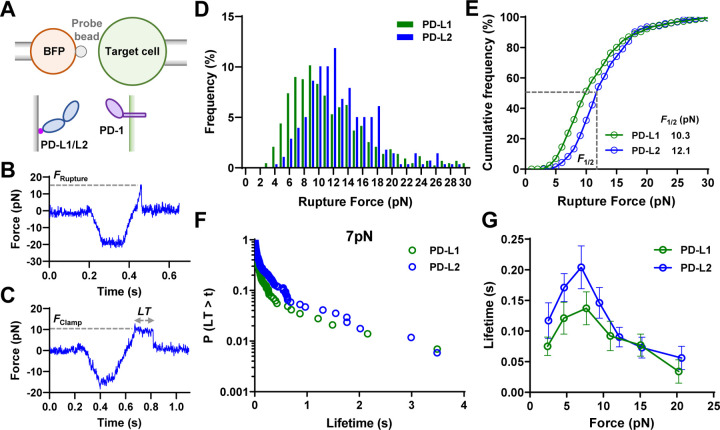
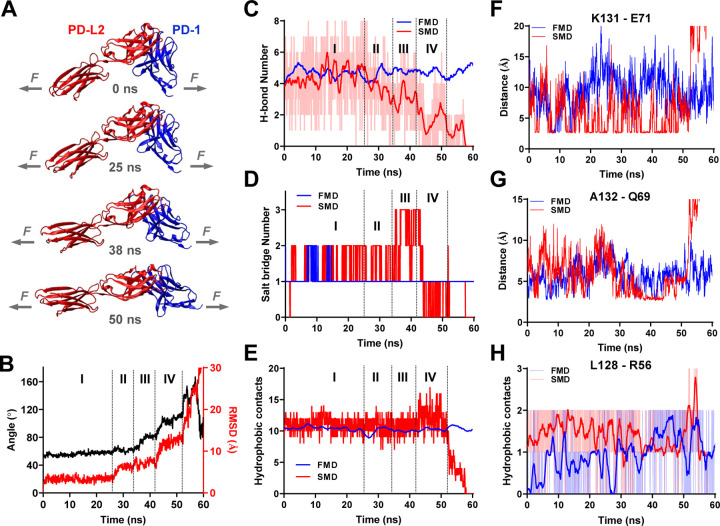
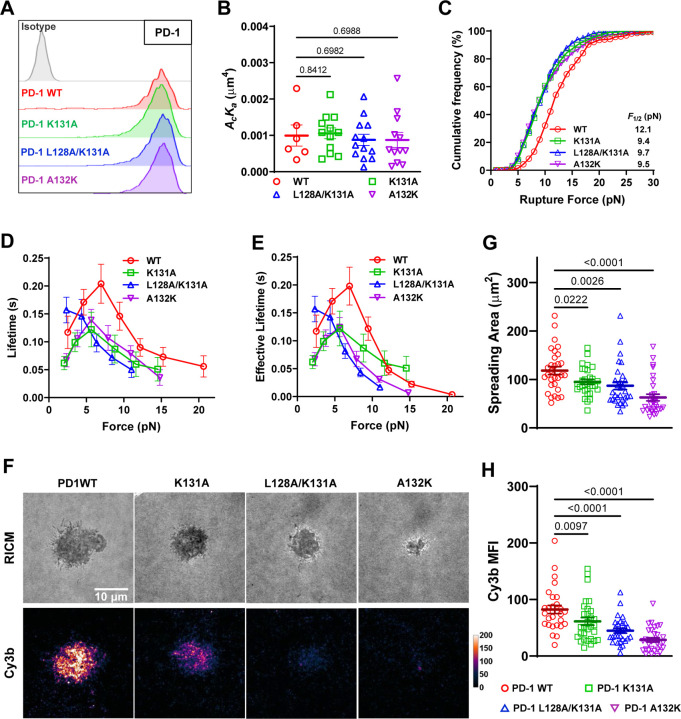
Immune checkpoint blockade targeting PD-1 shows great success in cancer therapy. However, the mechanism of how ligand binding initiates PD-1 signaling remains unclear. As prognosis markers of multiple cancers, soluble PD-L1 is found in patient sera and can bind PD-1, but fails to suppress T cell function. This and our previous observations that T cells exert endogenous forces on PD-1-PD-L2 bonds prompt the hypothesis that mechanical force might be critical to PD-1 triggering, which is missing in the soluble ligand case due to the lack of mechanical support afforded by surface-anchored ligand. Here we show that PD-1 function is eliminated or reduced when mechanical support on ligand is removed or dampened, respectively. Force spectroscopic analysis reveals that PD-1 forms catch bonds with both PD-Ligands <7 pN where force prolongs bond lifetime, but slip bonds >8 pN where force accelerates dissociation. Steered molecular dynamics finds PD-1-PD-L2 complex very sensitive to force due to the two molecules' "side-to-side" binding via β sheets. Pulling causes relative rotation and translation between the two molecules by stretching and aligning the complex along the force direction, yielding new atomic contacts not observed in the crystal structure. Compared to wild-type, PD-1 mutants targeting the force-induced new interactions maintain the same binding affinity but display lower rupture force, shorter bond lifetime, reduced tension, and most importantly, impaired capacity to suppress T cell activation. Our results uncover a mechanism for cells to probe the mechanical support of PD-1-PD-Ligand bonds using endogenous forces to regulate PD-1 triggering.
靶向程序性死亡受体1(PD-1)的免疫检查点阻断疗法在癌症治疗中取得了巨大成功。然而,配体结合启动PD-1信号传导的机制仍不清楚。作为多种癌症的预后标志物,可溶性程序性死亡配体1(PD-L1)在患者血清中被发现,并且可以结合PD-1,但无法抑制T细胞功能。这以及我们之前的观察结果,即T细胞对PD-1-PD-L2键施加内力,促使我们提出这样一个假设:机械力可能对PD-1的激活至关重要,而在可溶性配体的情况下,由于缺乏表面锚定配体提供的机械支持,这种机械力是缺失的。在这里,我们表明,当配体上的机械支持分别被去除或减弱时,PD-1的功能会被消除或降低。力谱分析表明,PD-1与两种PD配体在<7皮牛(pN)时形成捕获键,此时力会延长键的寿命,但在>8 pN时形成滑键,此时力会加速解离。定向分子动力学发现,由于两个分子通过β折叠“并排”结合,PD-1-PD-L2复合物对力非常敏感。拉伸会使复合物沿力的方向伸展和对齐,从而导致两个分子之间发生相对旋转和平移,产生晶体结构中未观察到的新原子接触。与野生型相比,靶向力诱导新相互作用的PD-1突变体保持相同的结合亲和力,但显示出更低的断裂力、更短的键寿命、更低的张力,最重要的是,抑制T细胞激活的能力受损。我们的结果揭示了一种细胞机制,即细胞利用内力探测PD-1-配体键的机械支持,以调节PD-1的激活。