Yi Jungseob, Lee Sangseon, Lim Sangsoo, Cho Changyun, Piao Yinhua, Yeo Marie, Kim Dongkyu, Kim Sun, Lee Sunho
Interdisciplinary Program in Artificial Intelligence, Seoul National University, Gwanak-ro 1, Gwanak-gu, Seoul, 08826, South Korea.
Institute of Computer Technology, Seoul National University, Gwanak-ro 1, Gwanak-gu, Seoul, 08826, South Korea.
Comput Struct Biotechnol J. 2023 Aug 25;21:4187-4195. doi: 10.1016/j.csbj.2023.08.016. eCollection 2023.
Lead identification is a fundamental step to prioritize candidate compounds for downstream drug discovery process. Machine learning (ML) and deep learning (DL) approaches are widely used to identify lead compounds using both chemical property and experimental information. However, ML or DL methods rarely consider compound similarity information directly since ML and DL models use abstract representation of molecules for model construction. Alternatively, data mining approaches are also used to explore chemical space with drug candidates by screening undesirable compounds. A major challenge for data mining approaches is to develop efficient data mining methods that search large chemical space for desirable lead compounds with low false positive rate.
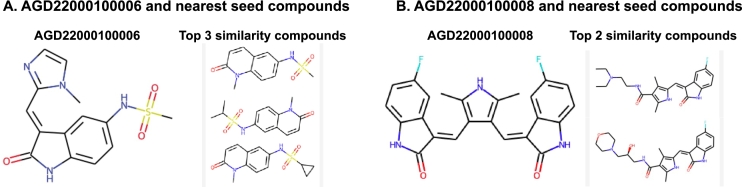
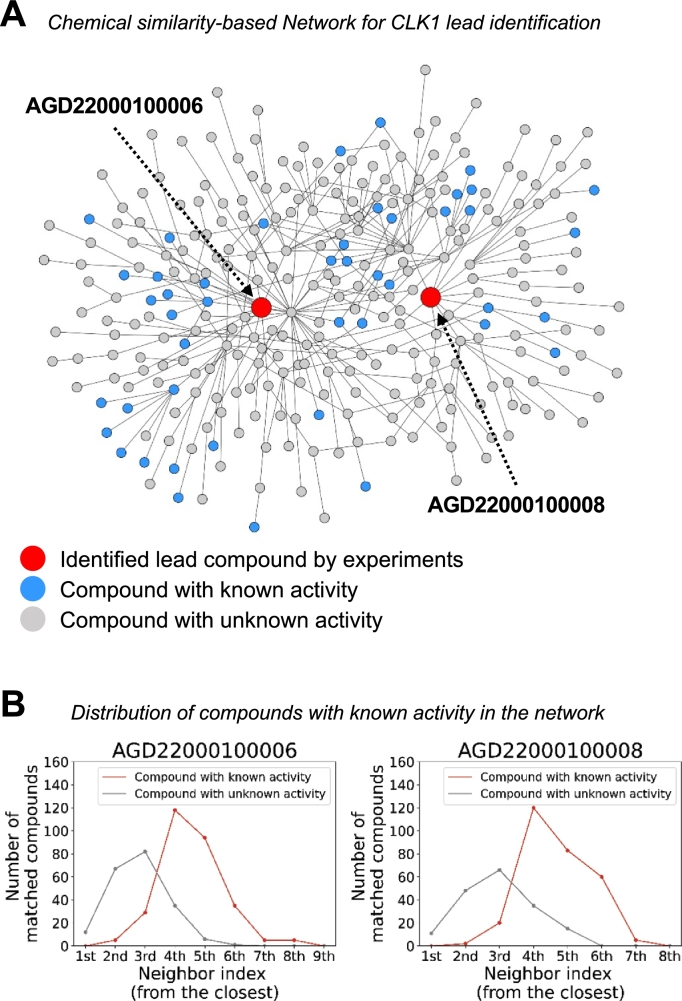
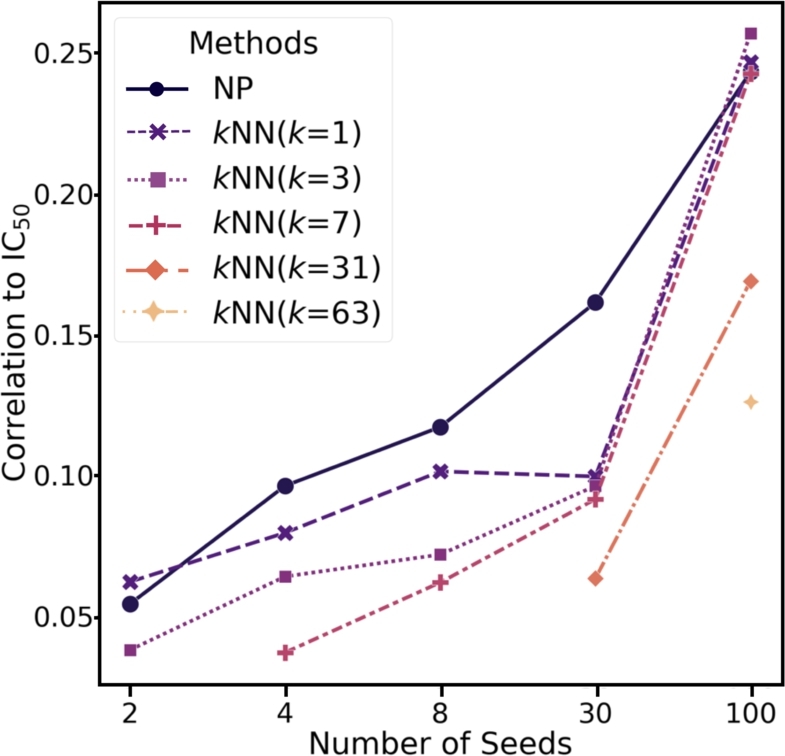
In this work, we developed a network propagation (NP) based data mining method for lead identification that performs search on an ensemble of chemical similarity networks. We compiled 14 fingerprint-based similarity networks. Given a target protein of interest, we use a deep learning-based drug target interaction model to narrow down compound candidates and then we use network propagation to prioritize drug candidates that are highly correlated with drug activity score such as IC. In an extensive experiment with BindingDB, we showed that our approach successfully discovered intentionally unlabeled compounds for given targets. To further demonstrate the prediction power of our approach, we identified 24 candidate leads for CLK1. Two out of five synthesizable candidates were experimentally validated in binding assays. In conclusion, our framework can be very useful for lead identification from very large compound databases such as ZINC.
先导化合物的识别是为下游药物发现过程确定候选化合物优先级的基本步骤。机器学习(ML)和深度学习(DL)方法被广泛用于利用化学性质和实验信息来识别先导化合物。然而,ML或DL方法很少直接考虑化合物相似性信息,因为ML和DL模型在构建模型时使用分子的抽象表示。另外,数据挖掘方法也被用于通过筛选不良化合物来探索含有候选药物的化学空间。数据挖掘方法面临的一个主要挑战是开发高效的数据挖掘方法,以便在大型化学空间中搜索具有低假阳性率的理想先导化合物。
在这项工作中,我们开发了一种基于网络传播(NP)的数据挖掘方法用于先导化合物识别,该方法在一组化学相似性网络上进行搜索。我们编制了14个基于指纹的相似性网络。给定一个感兴趣的目标蛋白,我们使用基于深度学习的药物-靶点相互作用模型来缩小候选化合物范围,然后使用网络传播来对与诸如IC等药物活性评分高度相关的候选药物进行优先级排序。在与BindingDB进行的广泛实验中,我们表明我们的方法成功发现了针对给定靶点的未标记化合物。为了进一步证明我们方法的预测能力,我们为CLK1识别了24个候选先导化合物。在可合成的五个候选化合物中,有两个在结合试验中得到了实验验证。总之,我们的框架对于从诸如ZINC这样的非常大的化合物数据库中识别先导化合物非常有用。