Hu Frank, He Francis, Yaron David J
Department of Chemistry, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, United States.
J Chem Theory Comput. 2023 Sep 26;19(18):6185-6196. doi: 10.1021/acs.jctc.3c00491. Epub 2023 Sep 13.
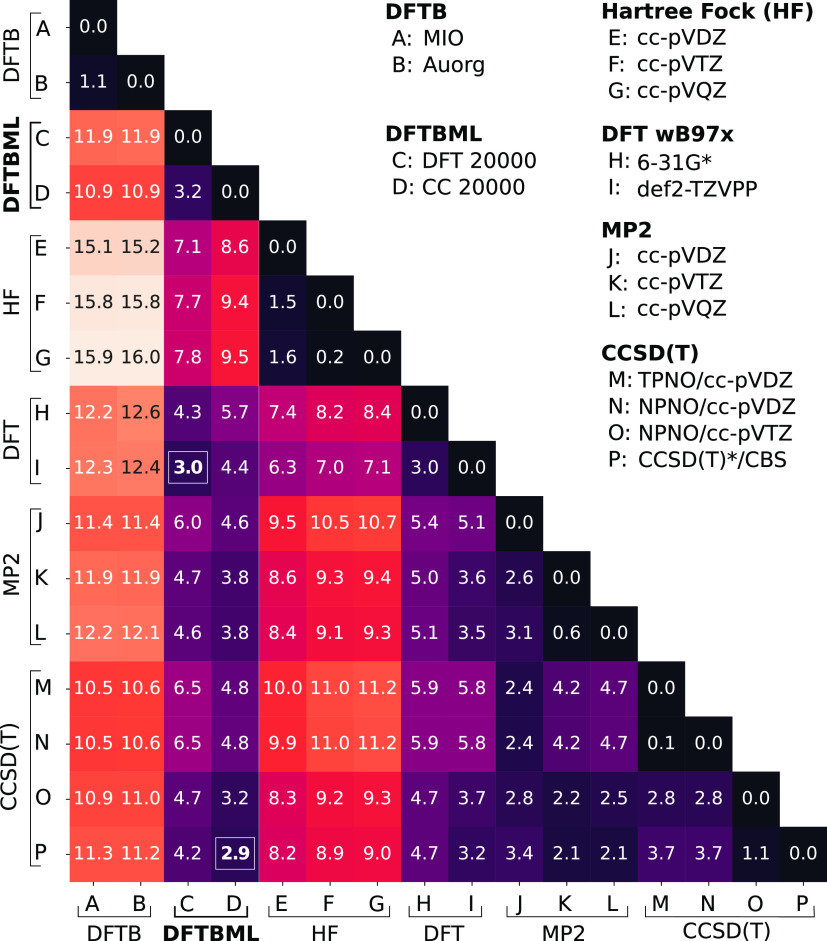
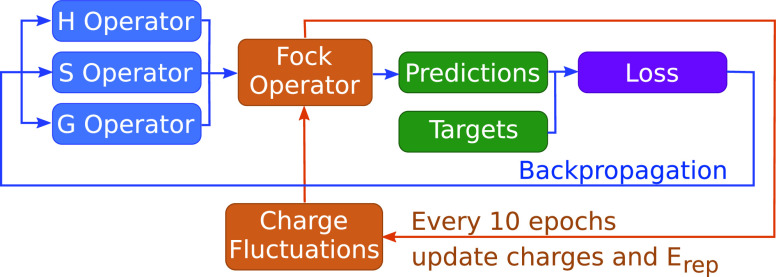
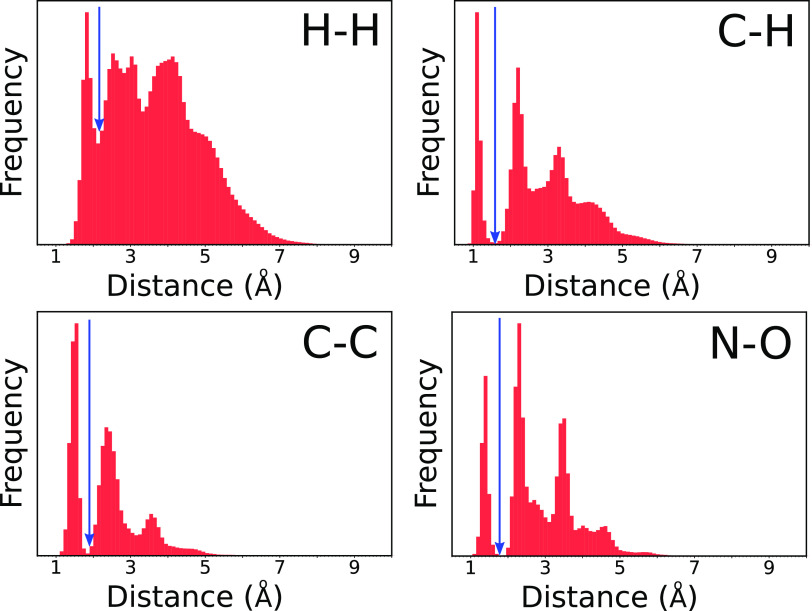
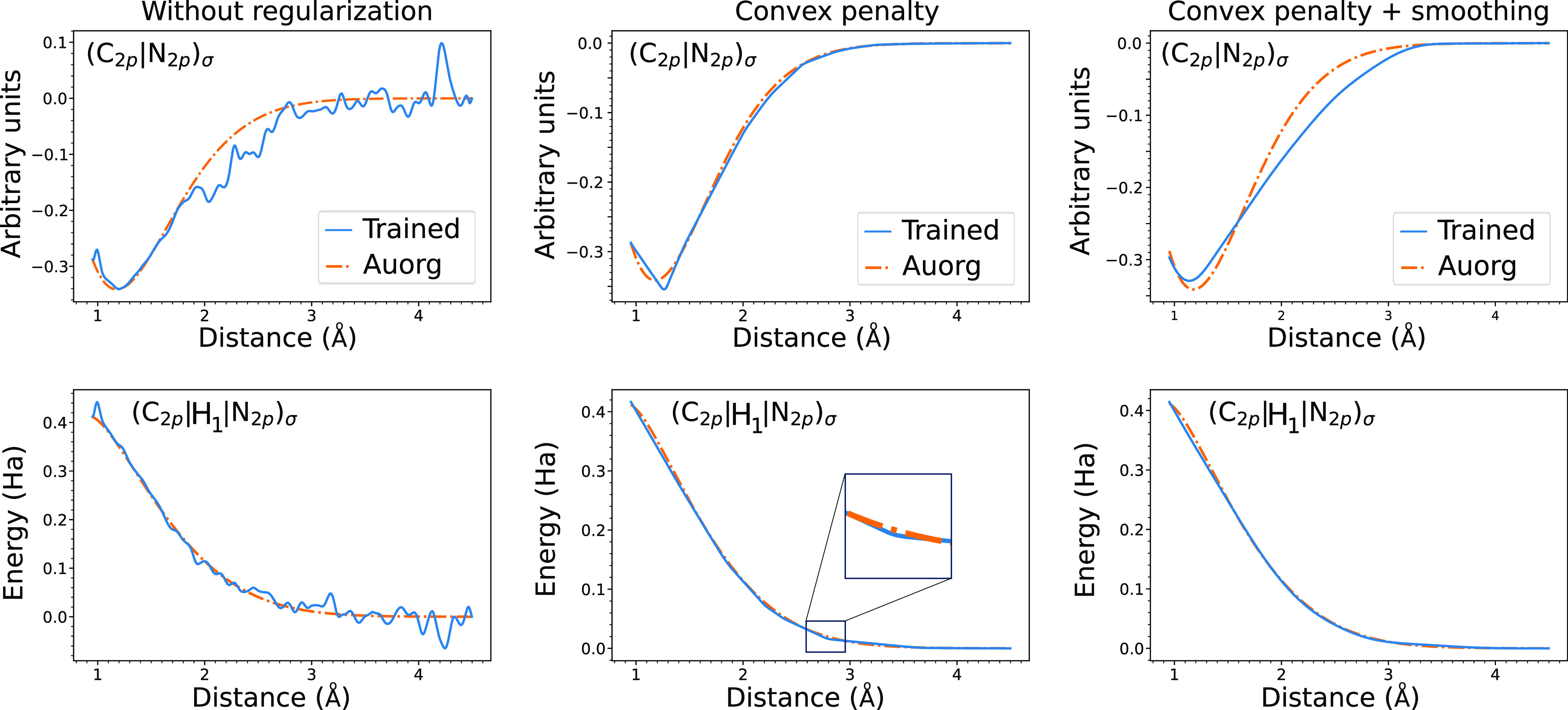
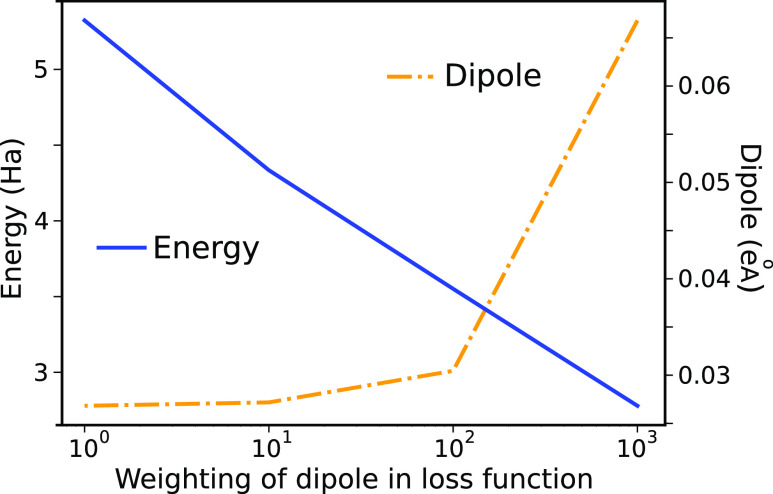
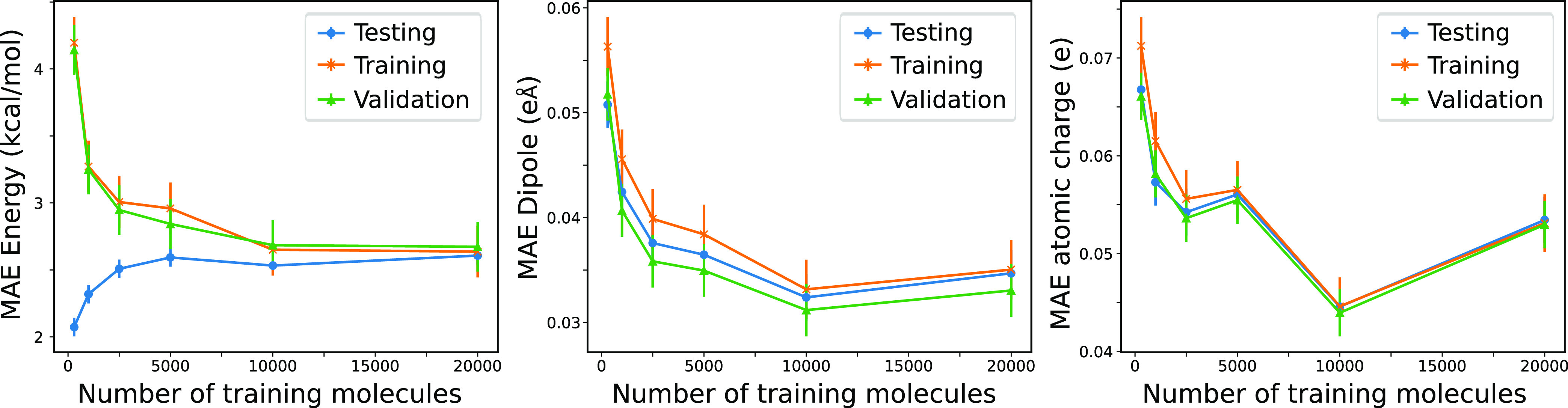
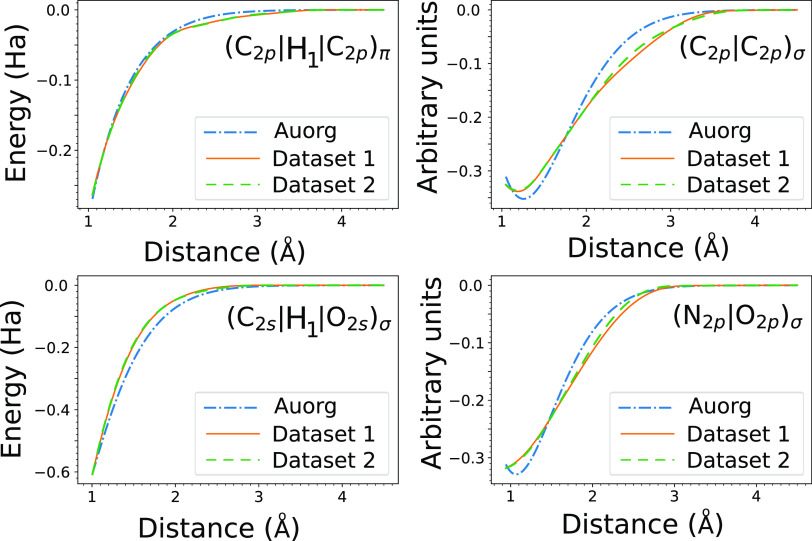
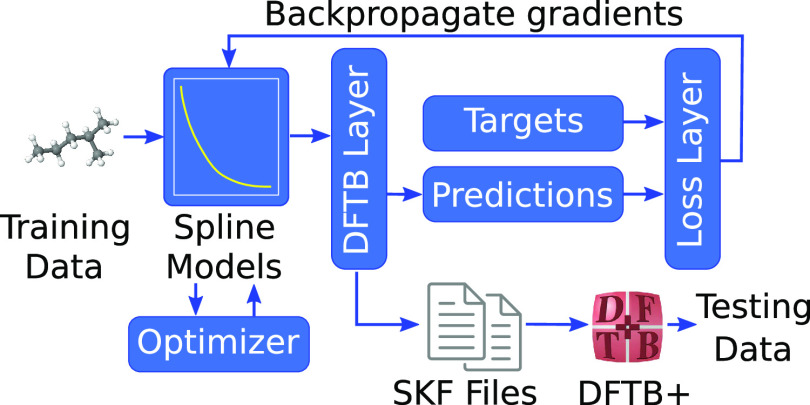
Quantum chemistry provides chemists with invaluable information, but the high computational cost limits the size and type of systems that can be studied. Machine learning (ML) has emerged as a means to dramatically lower the cost while maintaining high accuracy. However, ML models often sacrifice interpretability by using components such as the artificial neural networks of deep learning that function as black boxes. These components impart the flexibility needed to learn from large volumes of data but make it difficult to gain insight into the physical or chemical basis for the predictions. Here, we demonstrate that semiempirical quantum chemical (SEQC) models can learn from large volumes of data without sacrificing interpretability. The SEQC model is that of density-functional-based tight binding (DFTB) with fixed atomic orbital energies and interactions that are one-dimensional functions of the interatomic distance. This model is trained to data in a manner that is analogous to that used to train deep learning models. Using benchmarks that reflect the accuracy of the training data, we show that the resulting model maintains a physically reasonable functional form while achieving an accuracy, relative to coupled cluster energies with a complete basis set extrapolation (CCSD(T)*/CBS), that is comparable to that of density functional theory (DFT). This suggests that trained SEQC models can achieve a low computational cost and high accuracy without sacrificing interpretability. Use of a physically motivated model form also substantially reduces the amount of data needed to train the model compared to that required for deep learning models.
量子化学为化学家提供了极有价值的信息,但高昂的计算成本限制了可研究体系的规模和类型。机器学习(ML)已成为一种在保持高精度的同时大幅降低成本的手段。然而,ML模型通常会通过使用深度学习中的人工神经网络等作为黑箱的组件来牺牲可解释性。这些组件赋予了从大量数据中学习所需的灵活性,但却难以深入了解预测的物理或化学基础。在此,我们证明半经验量子化学(SEQC)模型可以在不牺牲可解释性的情况下从大量数据中学习。SEQC模型是基于密度泛函的紧束缚(DFTB)模型,其具有固定的原子轨道能量和作为原子间距离一维函数的相互作用。该模型以类似于训练深度学习模型的方式对数据进行训练。使用反映训练数据准确性的基准,我们表明所得模型在保持物理上合理的函数形式的同时,相对于具有完整基组外推的耦合簇能量(CCSD(T)*/CBS),实现了与密度泛函理论(DFT)相当的准确性。这表明经过训练的SEQC模型可以在不牺牲可解释性的情况下实现低计算成本和高精度。与深度学习模型相比,使用具有物理动机的模型形式还大幅减少了训练模型所需的数据量。