Institute for Quantitative Biomedicine, Rutgers-The State University of New Jersey, Piscataway, NJ 08854.
Department of Chemistry and Chemical Biology, Rutgers-The State University of New Jersey, Piscataway, NJ 08854.
Proc Natl Acad Sci U S A. 2023 Sep 26;120(39):e2303590120. doi: 10.1073/pnas.2303590120. Epub 2023 Sep 20.
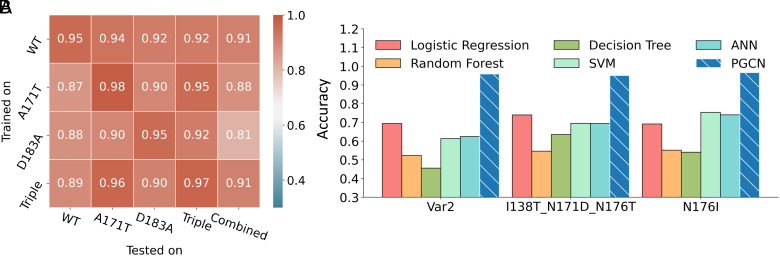
Site-specific proteolysis by the enzymatic cleavage of small linear sequence motifs is a key posttranslational modification involved in physiology and disease. The ability to robustly and rapidly predict protease-substrate specificity would also enable targeted proteolytic cleavage by designed proteases. Current methods for predicting protease specificity are limited to sequence pattern recognition in experimentally derived cleavage data obtained for libraries of potential substrates and generated separately for each protease variant. We reasoned that a more semantically rich and robust model of protease specificity could be developed by incorporating the energetics of molecular interactions between protease and substrates into machine learning workflows. We present Protein Graph Convolutional Network (PGCN), which develops a physically grounded, structure-based molecular interaction graph representation that describes molecular topology and interaction energetics to predict enzyme specificity. We show that PGCN accurately predicts the specificity landscapes of several variants of two model proteases. Node and edge ablation tests identified key graph elements for specificity prediction, some of which are consistent with known biochemical constraints for protease:substrate recognition. We used a pretrained PGCN model to guide the design of protease libraries for cleaving two noncanonical substrates, and found good agreement with experimental cleavage results. Importantly, the model can accurately assess designs featuring diversity at positions not present in the training data. The described methodology should enable the structure-based prediction of specificity landscapes of a wide variety of proteases and the construction of tailor-made protease editors for site-selectively and irreversibly modifying chosen target proteins.
位点特异性蛋白水解是一种涉及生理和疾病的关键翻译后修饰,通过酶对小线性序列基序的切割来实现。能够强大且快速地预测蛋白酶-底物特异性也将能够通过设计的蛋白酶进行靶向蛋白水解切割。目前预测蛋白酶特异性的方法仅限于从实验获得的潜在底物库的切割数据中进行序列模式识别,并且这些数据是为每个蛋白酶变体分别生成的。我们认为,可以通过将蛋白酶和底物之间分子相互作用的能量纳入机器学习工作流程,开发出更具语义丰富性和稳健性的蛋白酶特异性模型。我们提出了蛋白质图卷积网络(PGCN),它开发了一种基于物理的、基于结构的分子相互作用图表示形式,用于描述分子拓扑和相互作用能量,以预测酶特异性。我们表明,PGCN 可以准确预测两种模型蛋白酶的几个变体的特异性景观。节点和边消融测试确定了特异性预测的关键图元素,其中一些与蛋白酶:底物识别的已知生化约束一致。我们使用预训练的 PGCN 模型来指导针对两种非典型底物进行切割的蛋白酶文库的设计,并发现与实验切割结果具有良好的一致性。重要的是,该模型可以准确评估在训练数据中不存在的位置具有多样性的设计。所描述的方法学应该能够实现各种蛋白酶特异性景观的基于结构的预测,并构建用于选择性和不可逆地修饰选定靶蛋白的定制蛋白酶编辑器。