Department of Microbiology and Molecular Genetics, Institute for Biomedical Research Israel-Canada, Faculty of Medicine, The Hebrew University of Jerusalem, Jerusalem, Israel.
Nat Commun. 2022 Jan 10;13(1):176. doi: 10.1038/s41467-021-27838-9.
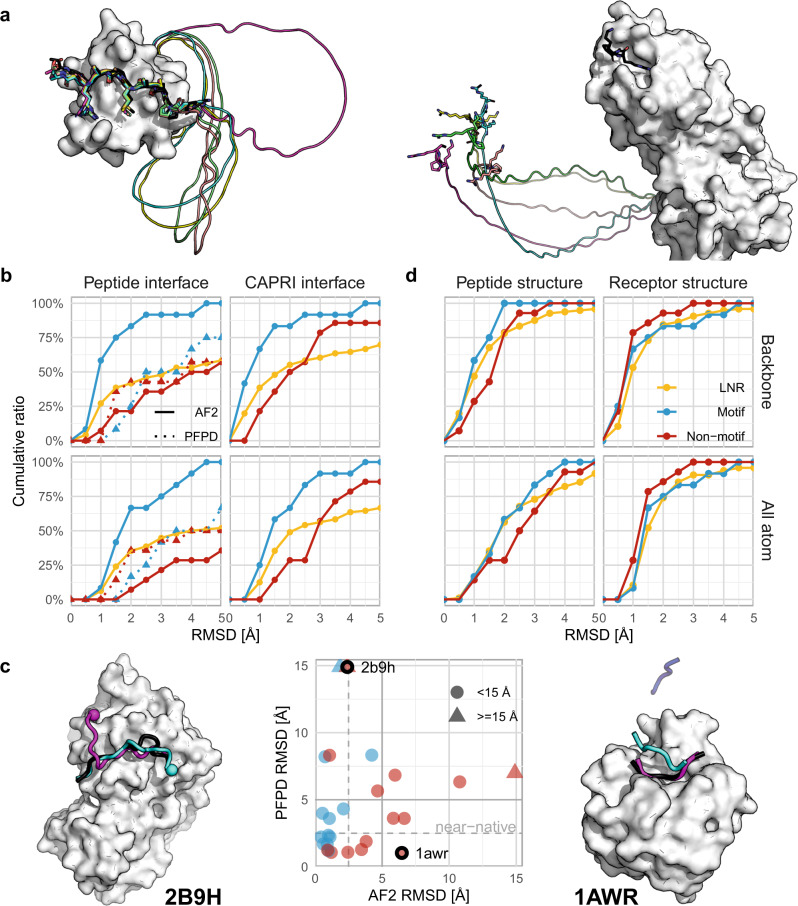
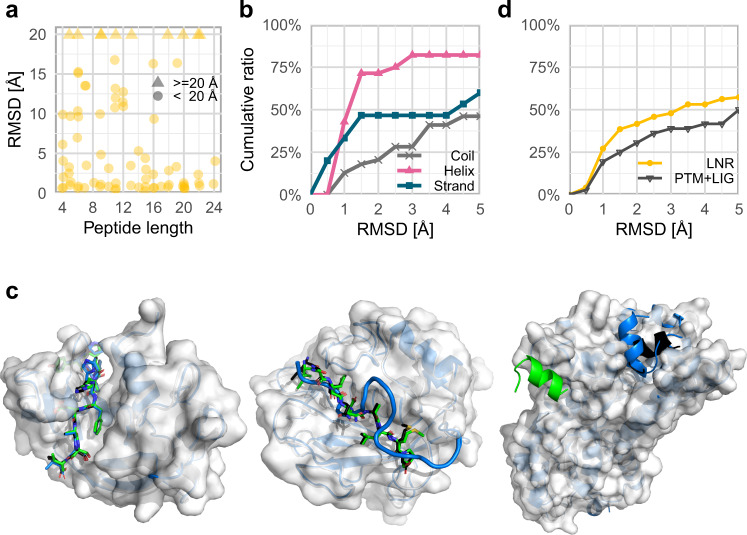
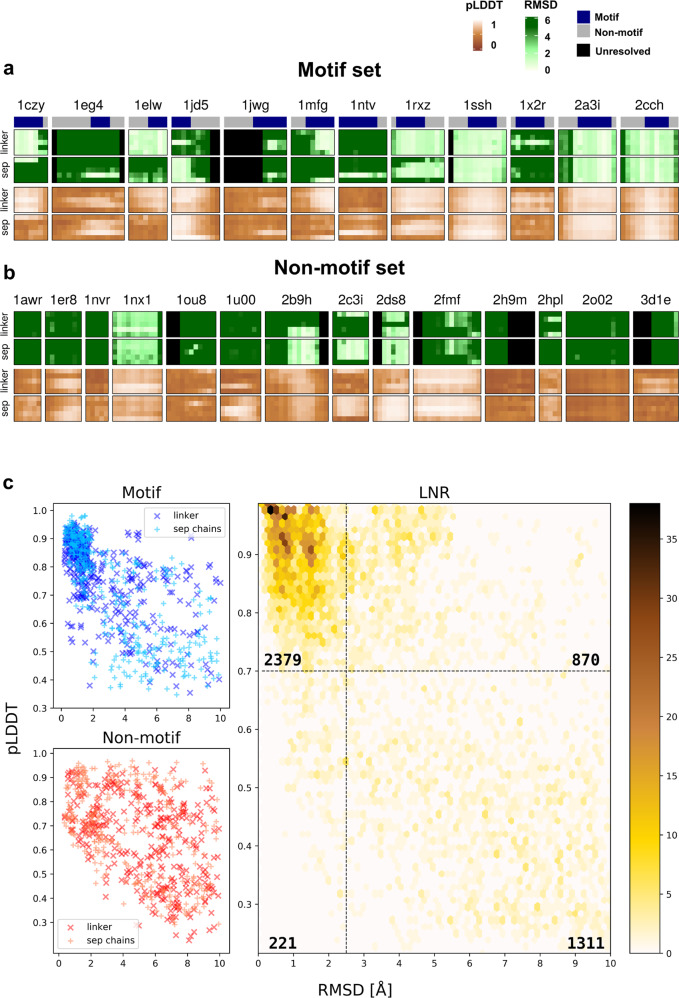
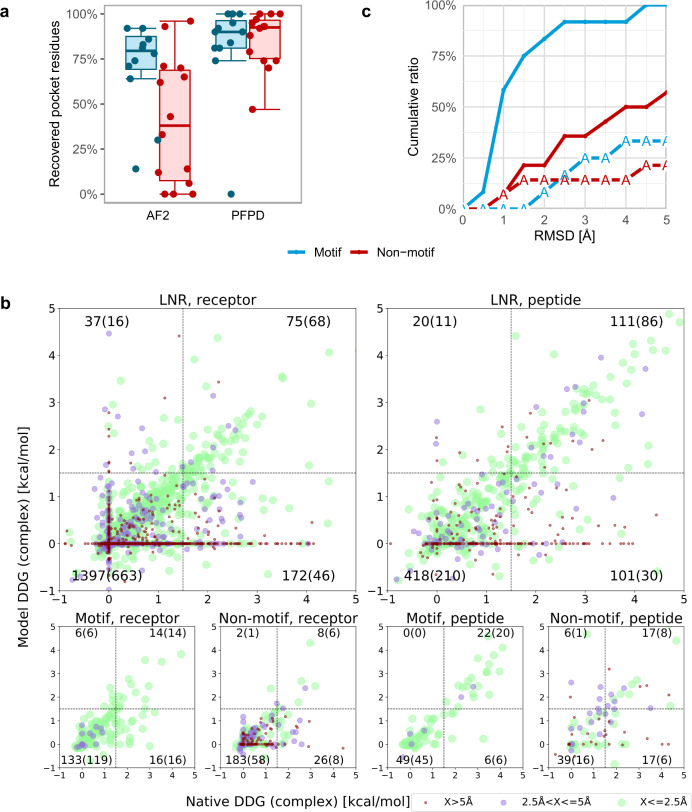
Highly accurate protein structure predictions by deep neural networks such as AlphaFold2 and RoseTTAFold have tremendous impact on structural biology and beyond. Here, we show that, although these deep learning approaches have originally been developed for the in silico folding of protein monomers, AlphaFold2 also enables quick and accurate modeling of peptide-protein interactions. Our simple implementation of AlphaFold2 generates peptide-protein complex models without requiring multiple sequence alignment information for the peptide partner, and can handle binding-induced conformational changes of the receptor. We explore what AlphaFold2 has memorized and learned, and describe specific examples that highlight differences compared to state-of-the-art peptide docking protocol PIPER-FlexPepDock. These results show that AlphaFold2 holds great promise for providing structural insight into a wide range of peptide-protein complexes, serving as a starting point for the detailed characterization and manipulation of these interactions.
深度学习方法(如 AlphaFold2 和 RoseTTAFold)能够实现高精度的蛋白质结构预测,对结构生物学乃至更广泛的领域都产生了巨大的影响。在这里,我们证明了这些深度学习方法虽然最初是为蛋白质单体的计算机折叠而开发的,但 AlphaFold2 也能够快速准确地模拟肽-蛋白质相互作用。我们对 AlphaFold2 的简单实现生成了肽-蛋白质复合物模型,而不需要肽伴侣的多重序列比对信息,并且可以处理受体的结合诱导构象变化。我们探索了 AlphaFold2 所记忆和学习的内容,并描述了一些具体的例子,这些例子突出了与最先进的肽对接协议 PIPER-FlexPepDock 的区别。这些结果表明,AlphaFold2 为提供广泛的肽-蛋白质复合物的结构见解提供了巨大的潜力,可作为详细表征和操作这些相互作用的起点。