Department of Anatomy and Cell Biology, Carver College of Medicine, University of Iowa, Iowa City, IA, USA.
Department of Environmental Health, Harvard T. H. Chan School of Public Health, Boston, MA, USA.
Nature. 2023 Sep;621(7980):857-867. doi: 10.1038/s41586-023-06549-9. Epub 2023 Sep 20.
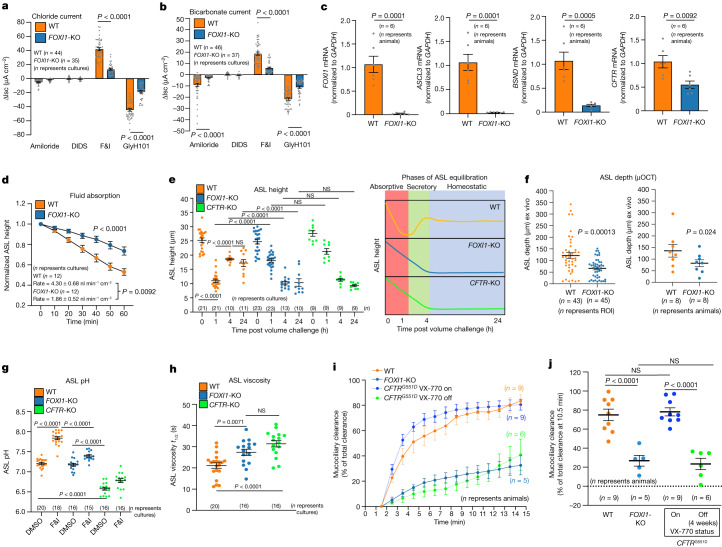
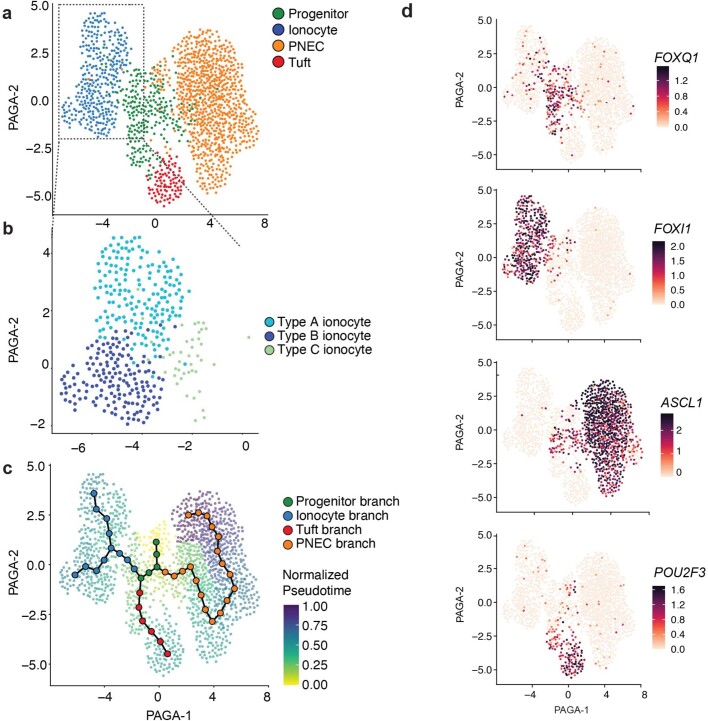
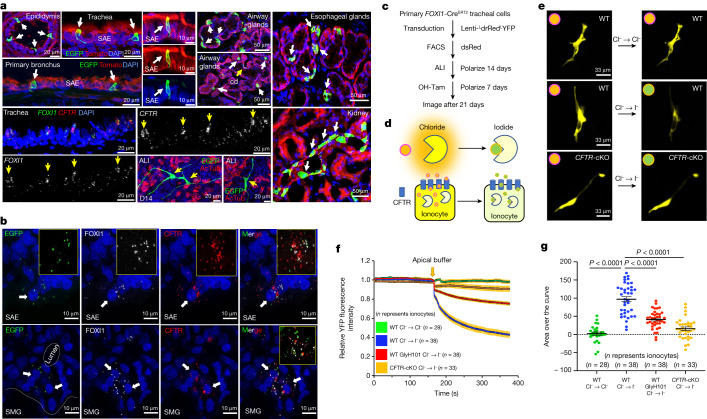
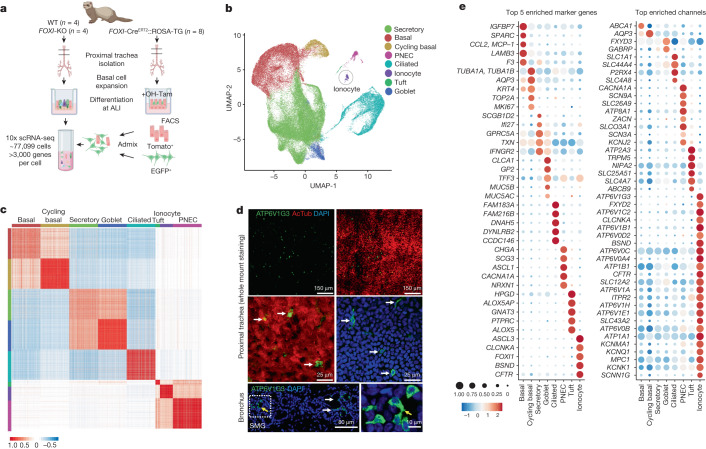
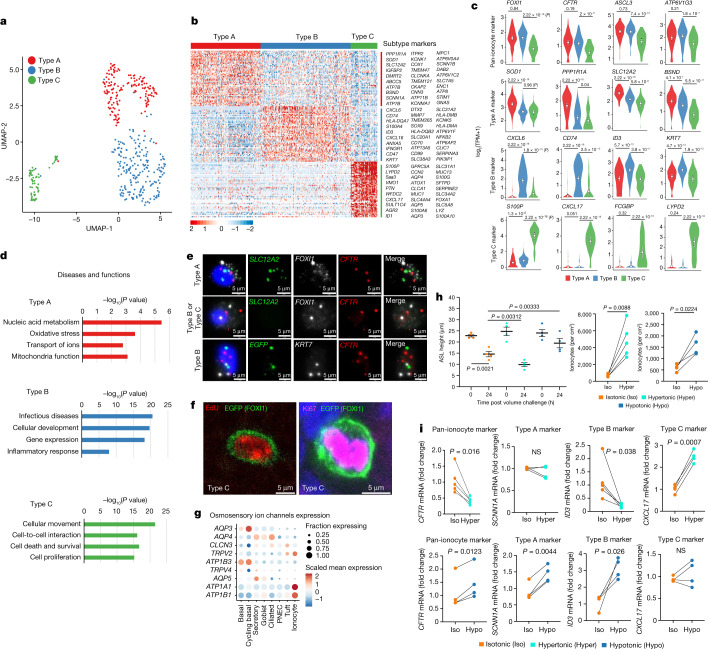
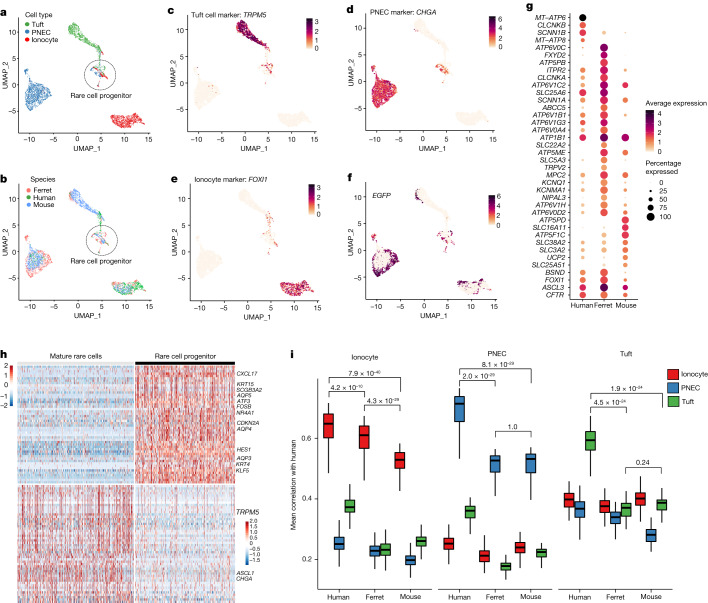
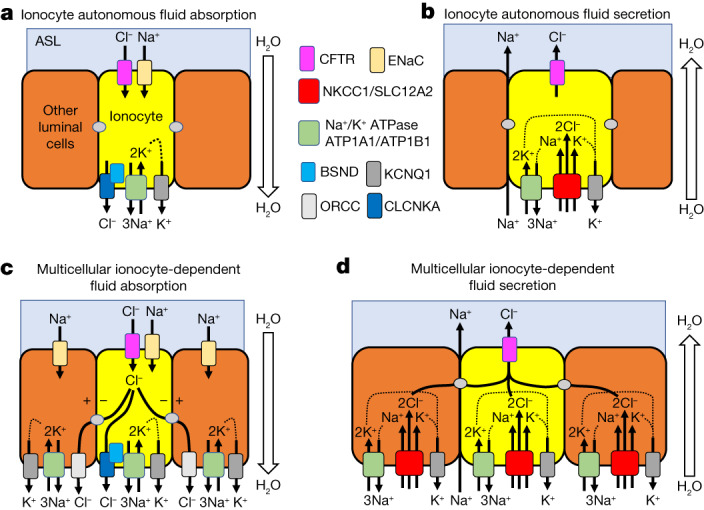
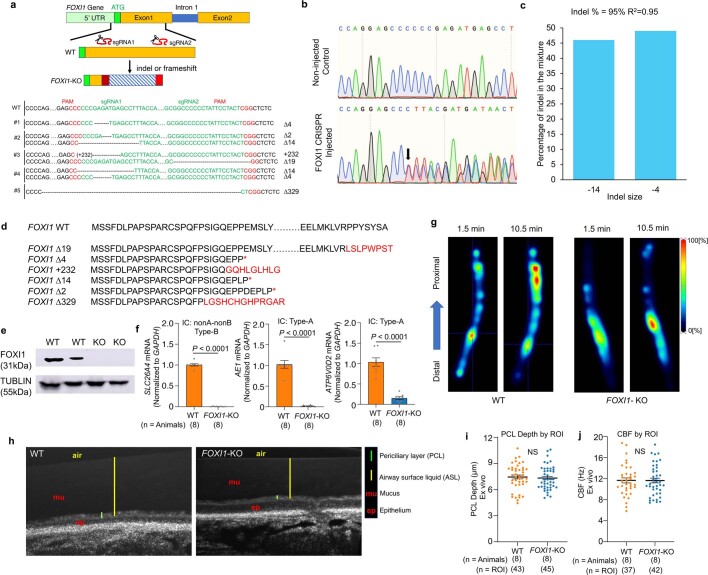
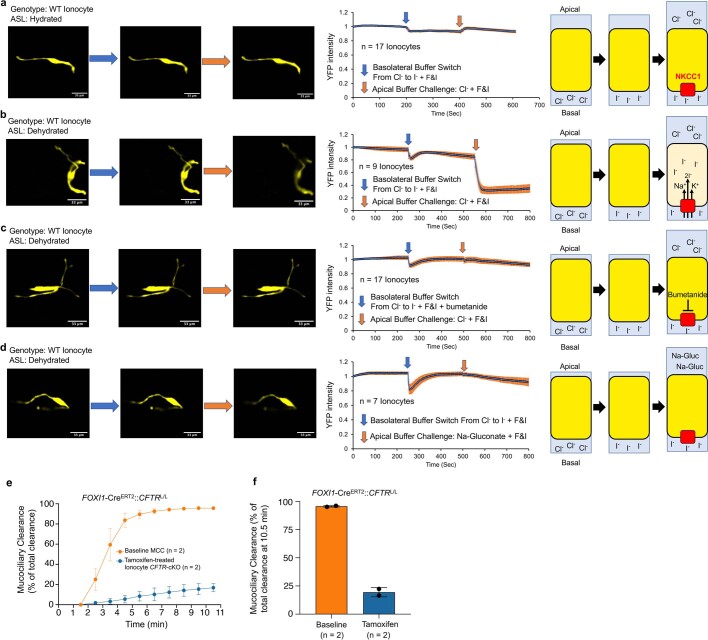
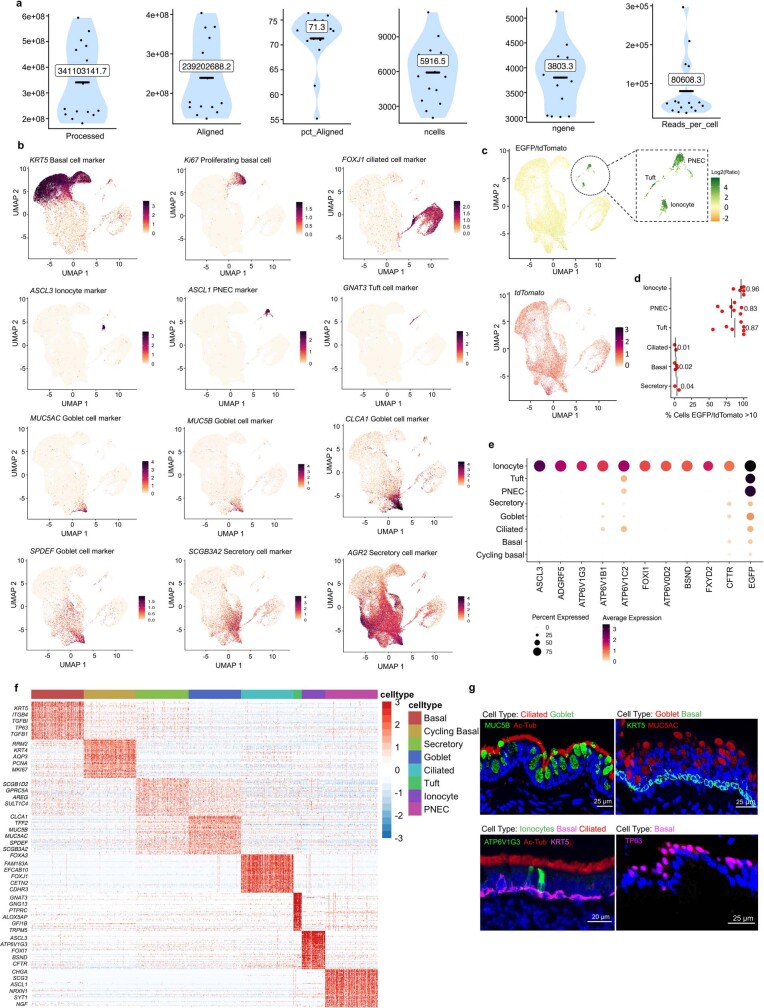
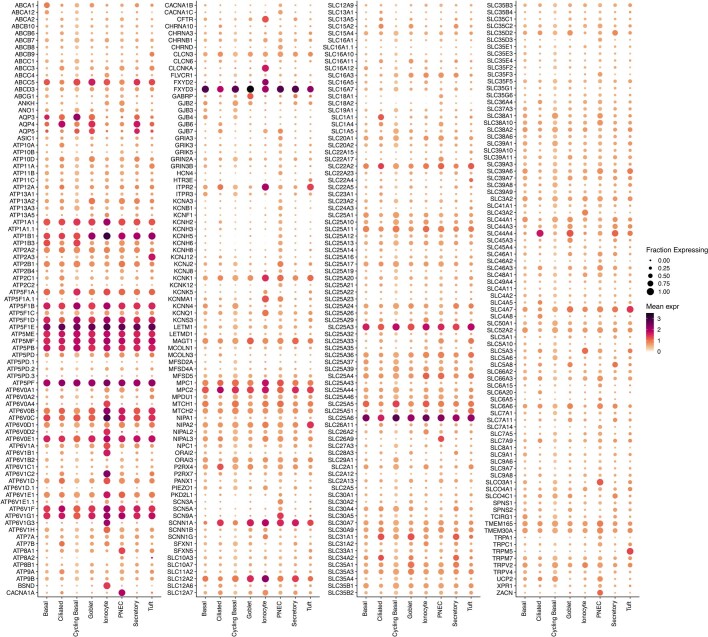
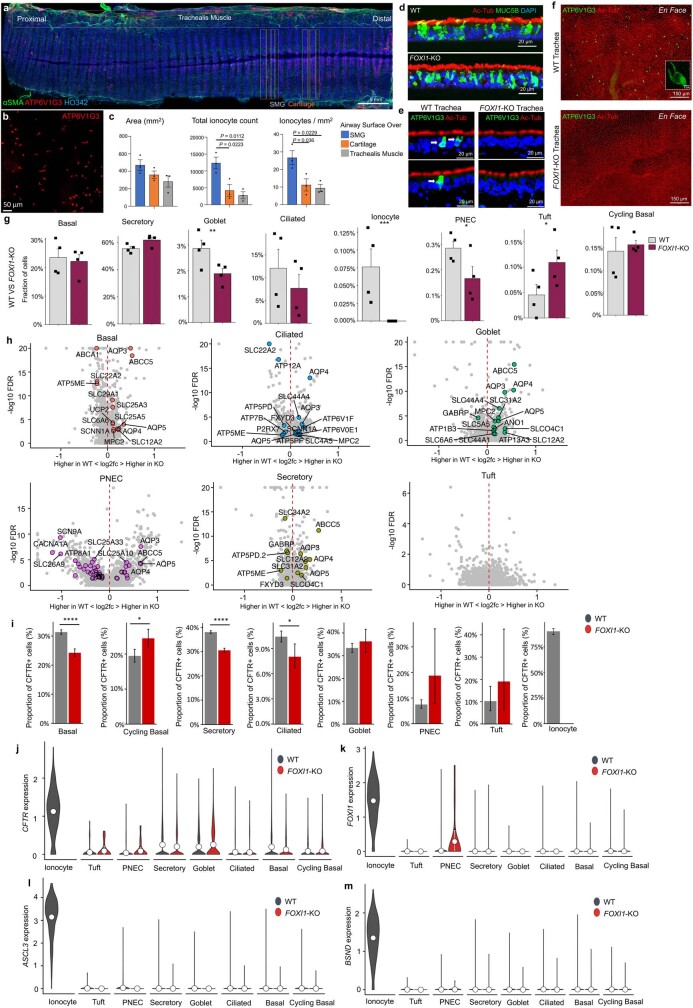
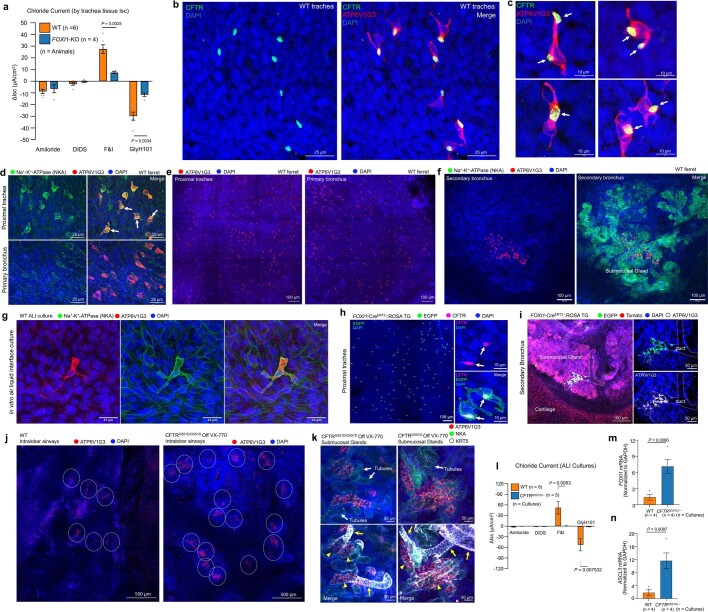
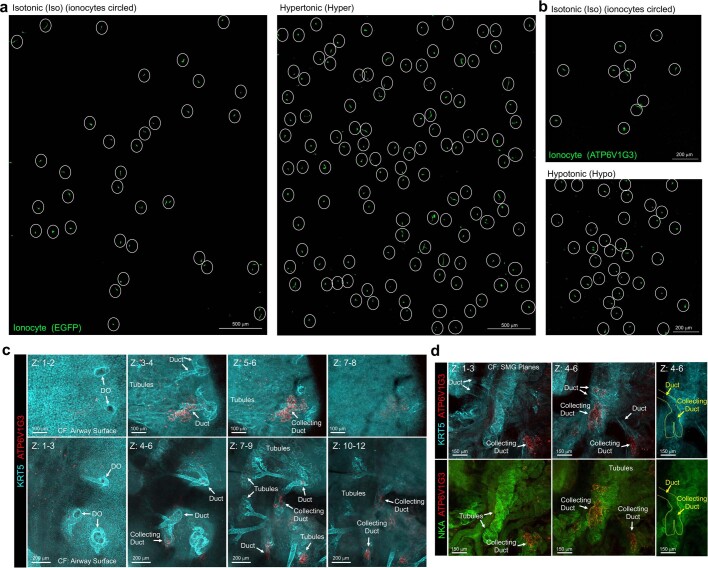
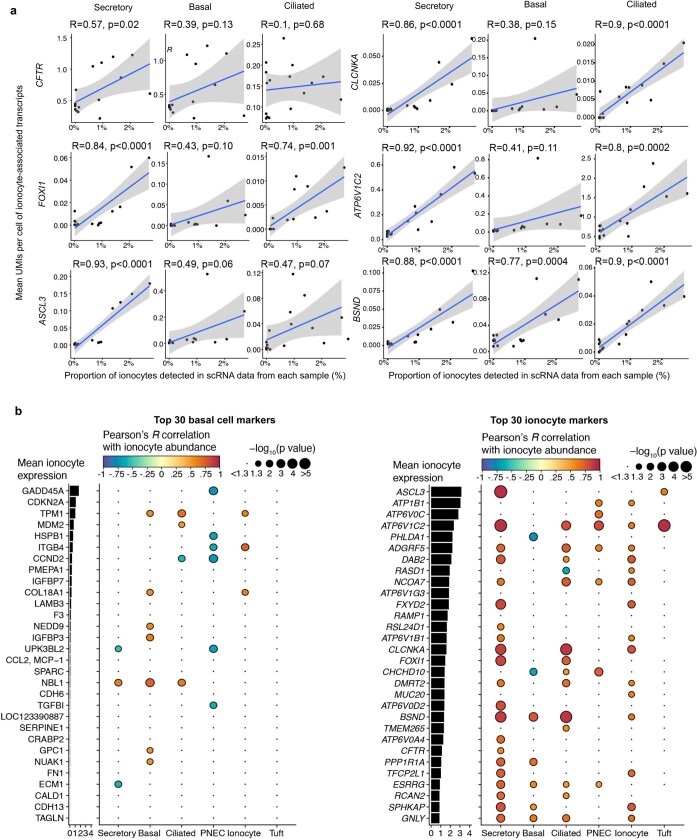
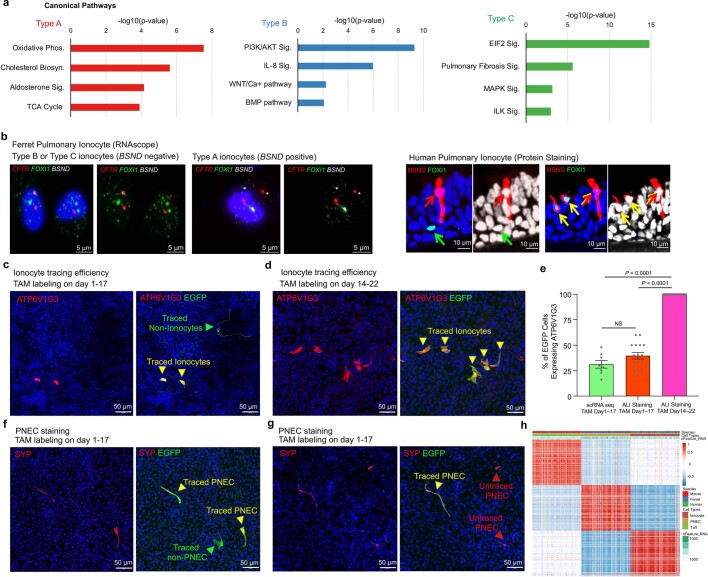
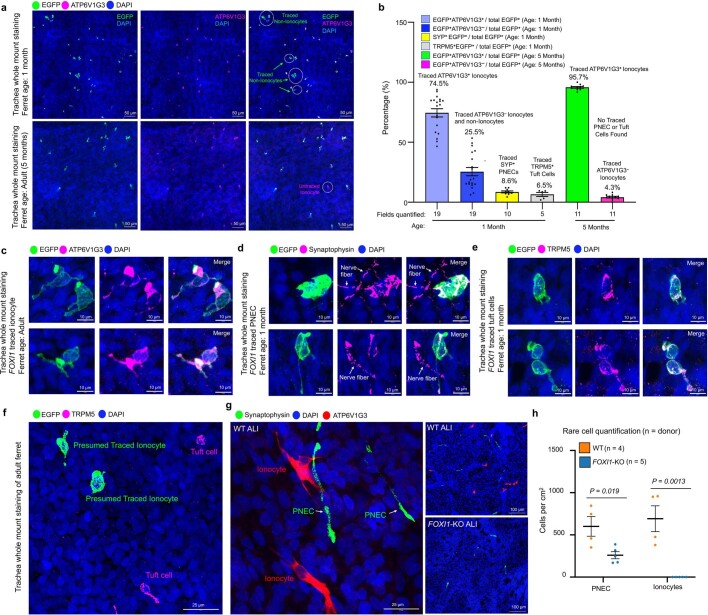
Speciation leads to adaptive changes in organ cellular physiology and creates challenges for studying rare cell-type functions that diverge between humans and mice. Rare cystic fibrosis transmembrane conductance regulator (CFTR)-rich pulmonary ionocytes exist throughout the cartilaginous airways of humans, but limited presence and divergent biology in the proximal trachea of mice has prevented the use of traditional transgenic models to elucidate ionocyte functions in the airway. Here we describe the creation and use of conditional genetic ferret models to dissect pulmonary ionocyte biology and function by enabling ionocyte lineage tracing (FOXI1-Cre::ROSA-TG), ionocyte ablation (FOXI1-KO) and ionocyte-specific deletion of CFTR (FOXI1-Cre::CFTR). By comparing these models with cystic fibrosis ferrets, we demonstrate that ionocytes control airway surface liquid absorption, secretion, pH and mucus viscosity-leading to reduced airway surface liquid volume and impaired mucociliary clearance in cystic fibrosis, FOXI1-KO and FOXI1-Cre::CFTR ferrets. These processes are regulated by CFTR-dependent ionocyte transport of Cl and HCO. Single-cell transcriptomics and in vivo lineage tracing revealed three subtypes of pulmonary ionocytes and a FOXI1-lineage common rare cell progenitor for ionocytes, tuft cells and neuroendocrine cells during airway development. Thus, rare pulmonary ionocytes perform critical CFTR-dependent functions in the proximal airway that are hallmark features of cystic fibrosis airway disease. These studies provide a road map for using conditional genetics in the first non-rodent mammal to address gene function, cell biology and disease processes that have greater evolutionary conservation between humans and ferrets.
物种形成导致器官细胞生理学的适应性变化,并为研究人类和小鼠之间存在分歧的罕见细胞类型功能带来挑战。在人类的软骨气道中存在丰富的囊性纤维化跨膜电导调节剂 (CFTR) 的罕见离子细胞,但在小鼠的近段气管中,其存在有限且生物学特性不同,这使得传统的转基因模型无法用于阐明气道中离子细胞的功能。在这里,我们描述了通过启用离子细胞谱系追踪 (FOXI1-Cre::ROSA-TG)、离子细胞消融 (FOXI1-KO) 和 CFTR 的离子细胞特异性缺失 (FOXI1-Cre::CFTR),创建和使用条件性遗传雪貂模型来剖析肺离子细胞生物学和功能。通过将这些模型与囊性纤维化雪貂进行比较,我们证明离子细胞控制气道表面液体的吸收、分泌、pH 值和粘液粘度,导致囊性纤维化、FOXI1-KO 和 FOXI1-Cre::CFTR 雪貂的气道表面液体体积减少和粘液纤毛清除功能受损。这些过程受 CFTR 依赖性离子细胞 Cl 和 HCO 转运调节。单细胞转录组学和体内谱系追踪揭示了三种肺离子细胞亚型,以及在气道发育过程中,FOXI1 谱系的离子细胞、微绒毛细胞和神经内分泌细胞的共同罕见细胞前体。因此,罕见的肺离子细胞在近端气道中执行关键的 CFTR 依赖性功能,这是囊性纤维化气道疾病的标志性特征。这些研究为在第一个非啮齿类哺乳动物中使用条件性遗传学提供了路线图,以解决基因功能、细胞生物学和疾病过程,这些在人类和雪貂之间具有更大的进化保守性。