Department of Computer Science, University of Tsukuba, Tsukuba 3058577, Japan.
Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu, China.
Bioinformatics. 2023 Nov 1;39(11). doi: 10.1093/bioinformatics/btad627.
The rapid and extensive transmission of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has led to an unprecedented global health emergency, affecting millions of people and causing an immense socioeconomic impact. The identification of SARS-CoV-2 phosphorylation sites plays an important role in unraveling the complex molecular mechanisms behind infection and the resulting alterations in host cell pathways. However, currently available prediction tools for identifying these sites lack accuracy and efficiency.
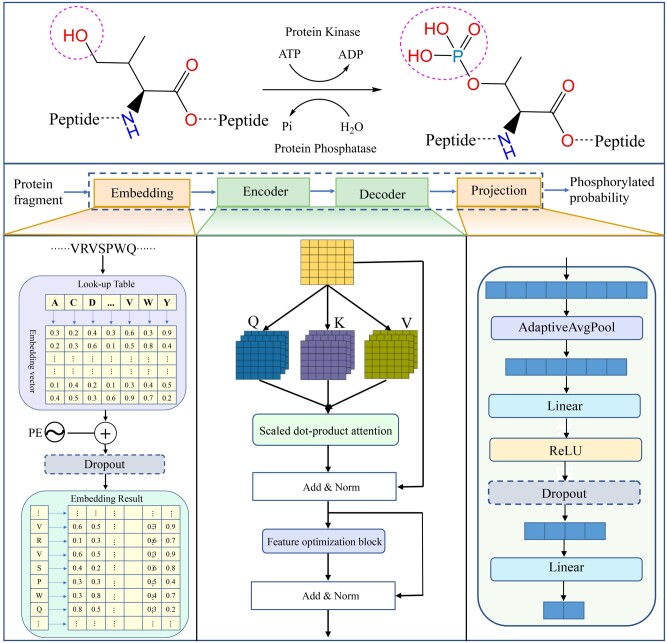
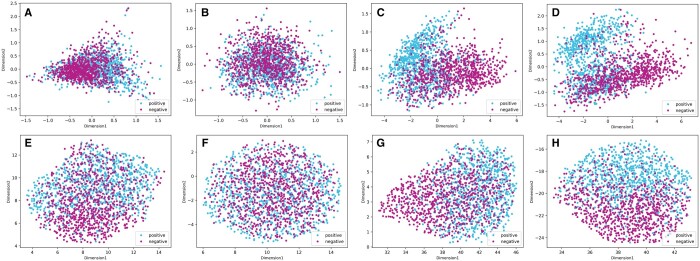
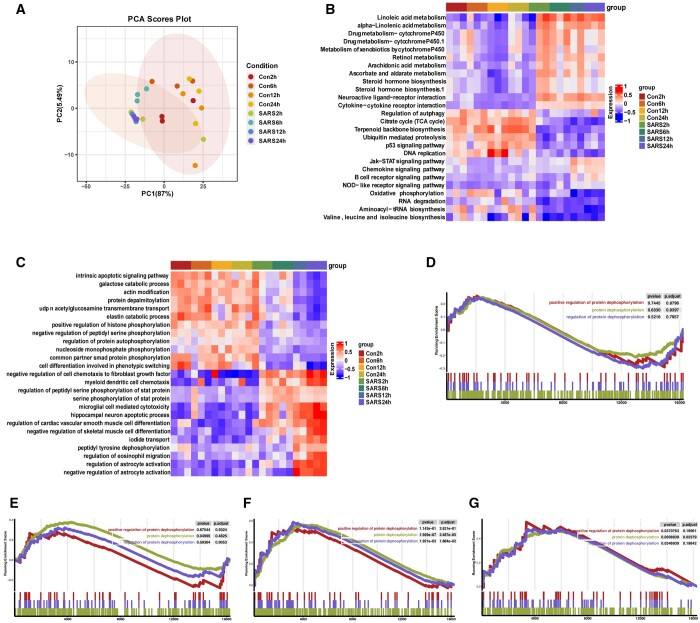
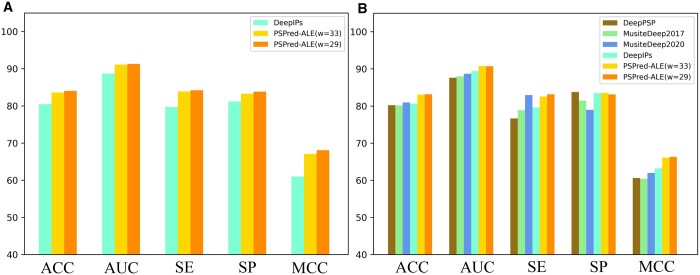
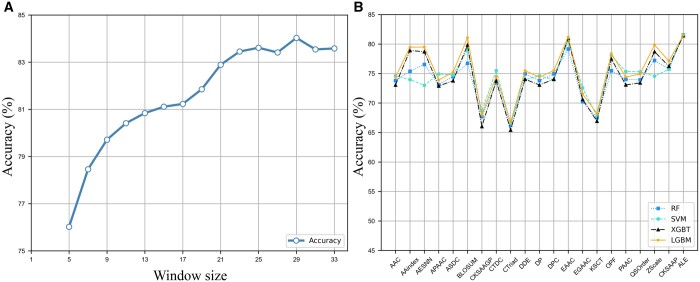
In this study, we presented a comprehensive biological function analysis of SARS-CoV-2 infection in a clonal human lung epithelial A549 cell, revealing dramatic changes in protein phosphorylation pathways in host cells. Moreover, a novel deep learning predictor called PSPred-ALE is specifically designed to identify phosphorylation sites in human host cells that are infected with SARS-CoV-2. The key idea of PSPred-ALE lies in the use of a self-adaptive learning embedding algorithm, which enables the automatic extraction of context sequential features from protein sequences. In addition, the tool uses multihead attention module that enables the capturing of global information, further improving the accuracy of predictions. Comparative analysis of features demonstrated that the self-adaptive learning embedding features are superior to hand-crafted statistical features in capturing discriminative sequence information. Benchmarking comparison shows that PSPred-ALE outperforms the state-of-the-art prediction tools and achieves robust performance. Therefore, the proposed model can effectively identify phosphorylation sites assistant the biomedical scientists in understanding the mechanism of phosphorylation in SARS-CoV-2 infection.
PSPred-ALE is available at https://github.com/jiaoshihu/PSPred-ALE and Zenodo (https://doi.org/10.5281/zenodo.8330277).
严重急性呼吸系统综合症冠状病毒 2(SARS-CoV-2)的快速广泛传播导致了一场前所未有的全球卫生紧急事件,影响了数百万人,并造成了巨大的社会经济影响。鉴定 SARS-CoV-2 的磷酸化位点在揭示感染背后的复杂分子机制以及宿主细胞途径的改变方面发挥着重要作用。然而,目前用于识别这些位点的预测工具缺乏准确性和效率。
在这项研究中,我们对克隆人肺上皮 A549 细胞中 SARS-CoV-2 的感染进行了全面的生物学功能分析,揭示了宿主细胞中蛋白质磷酸化途径的巨大变化。此外,我们专门设计了一种名为 PSPred-ALE 的新型深度学习预测器,用于识别感染 SARS-CoV-2 的人宿主细胞中的磷酸化位点。PSPred-ALE 的关键思想在于使用自适应学习嵌入算法,该算法能够自动从蛋白质序列中提取上下文序列特征。此外,该工具还使用多头注意力模块,能够捕获全局信息,从而进一步提高预测的准确性。对特征的比较分析表明,自适应学习嵌入特征在捕获区分性序列信息方面优于手工制作的统计特征。基准比较表明,PSPred-ALE 优于最先进的预测工具,并实现了稳健的性能。因此,该模型可以有效地识别磷酸化位点,协助生物医学科学家理解 SARS-CoV-2 感染中的磷酸化机制。
PSPred-ALE 可在 https://github.com/jiaoshihu/PSPred-ALE 和 Zenodo(https://doi.org/10.5281/zenodo.8330277)上获得。