Department of Orthopedics, The Second Affiliated Hospital of Harbin Medical University, Harbin, China.
Front Immunol. 2023 Oct 4;14:1202758. doi: 10.3389/fimmu.2023.1202758. eCollection 2023.
Osteoarthritis (OA) progression involves multiple factors, including cartilage erosion as the basic pathological mechanism of degeneration, and is closely related to chondrocyte apoptosis. To analyze the correlation between apoptosis and OA development, we selected apoptosis genes from the differentially expressed genes (DEGs) between OA and normal samples from the Gene Expression Omnibus (GEO) database, used lasso regression analysis to identify characteristic genes, and performed consensus cluster analysis to further explore the pathogenesis of this disease.
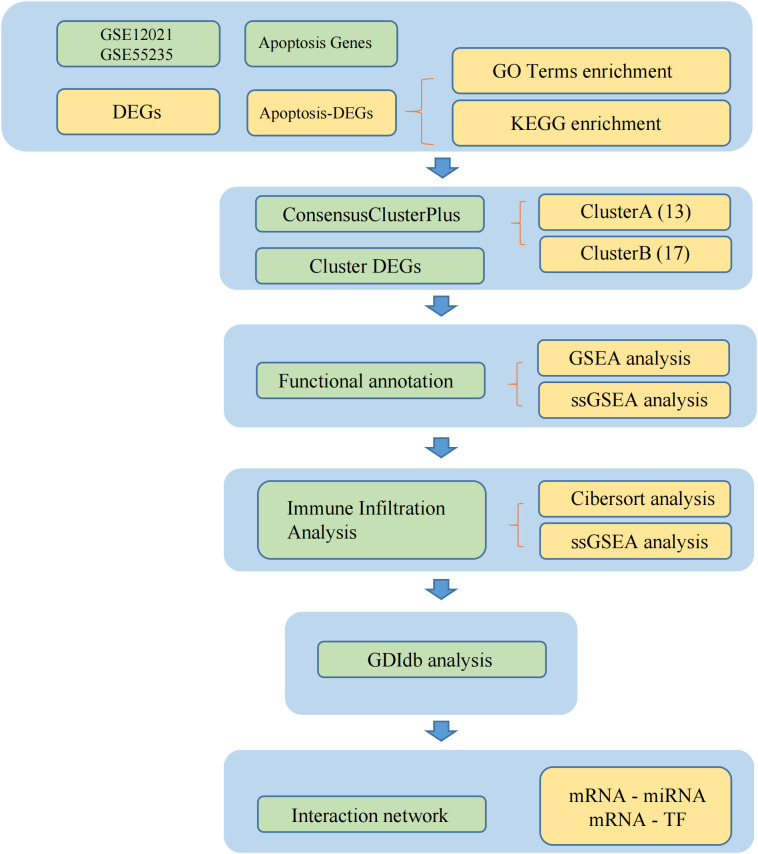
The Gene expression profile datasets of OA samples, GSE12021 and GSE55235, were downloaded from GEO. The datasets were combined and analyzed for DEGs. Apoptosis-related genes (ARGs) were collected from the GeneCards database and intersected with DEGs for apoptosis-related DEGs (ARDEGs). Least absolute shrinkage and selection operator (LASSO) regression analysis was performed to obtain characteristic genes, and a nomogram was constructed based on these genes. A consensus cluster analysis was performed to divide the patients into clusters. The immune characteristics, functional enrichment, and immune infiltration statuses of the clusters were compared. In addition, a protein-protein interaction network of mRNA drugs, mRNA-transcription factors (TFs), and mRNA-miRNAs was constructed.
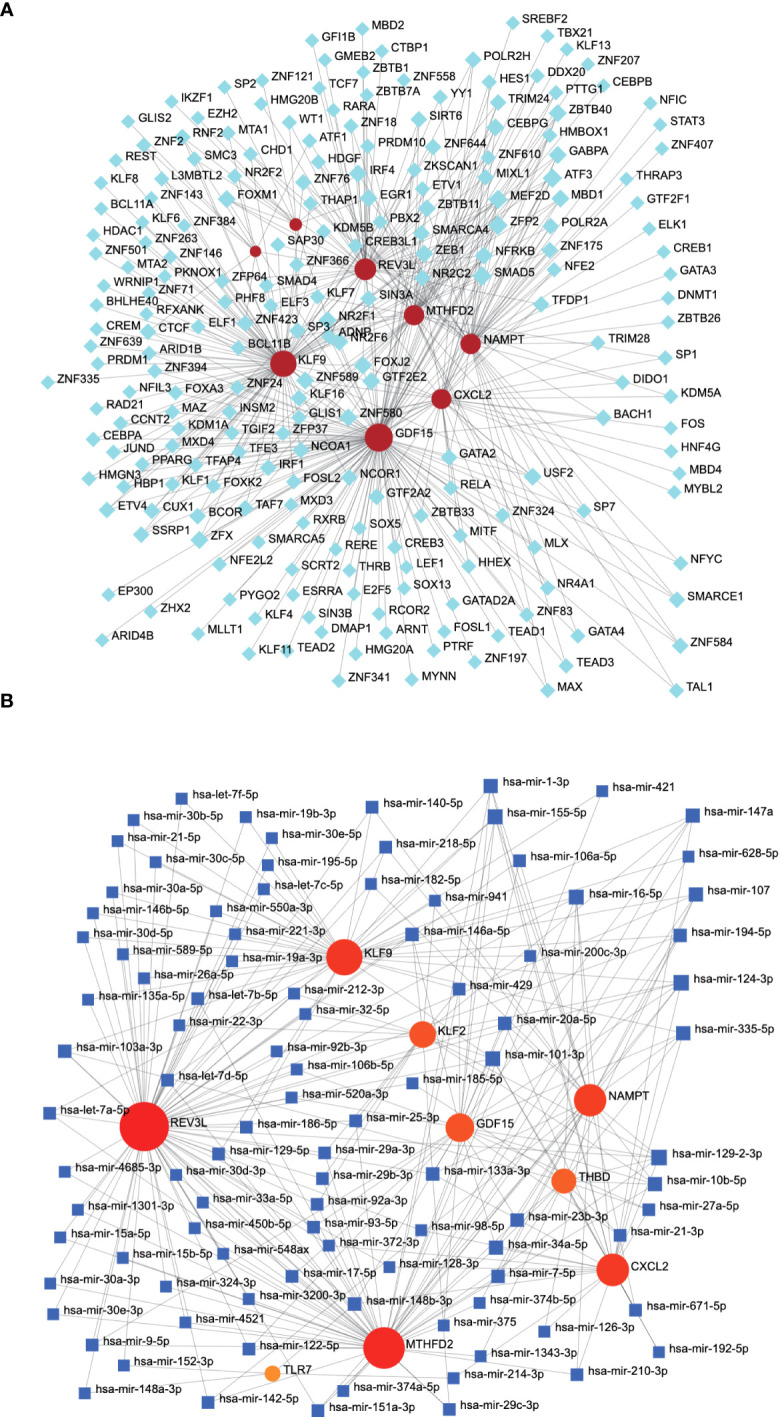
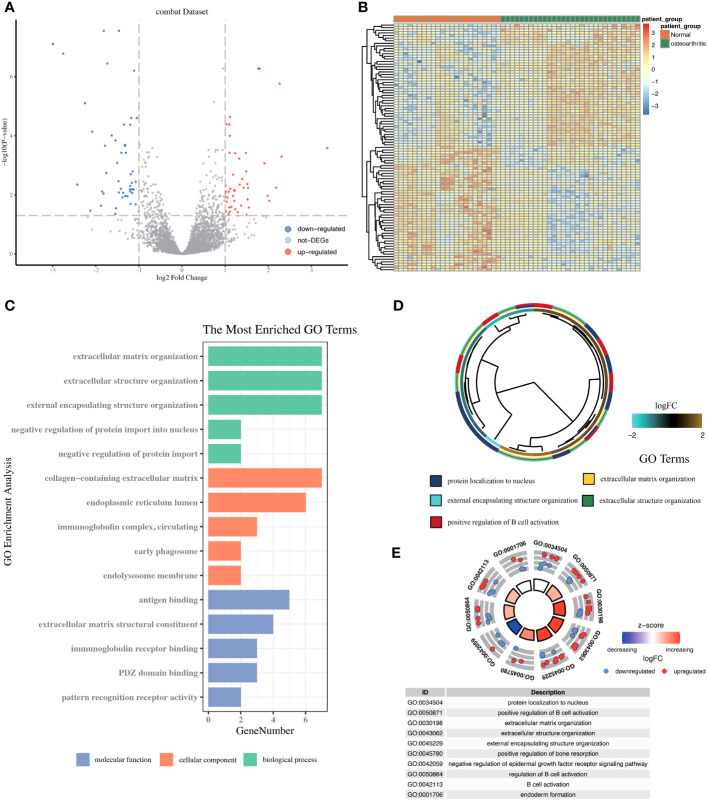
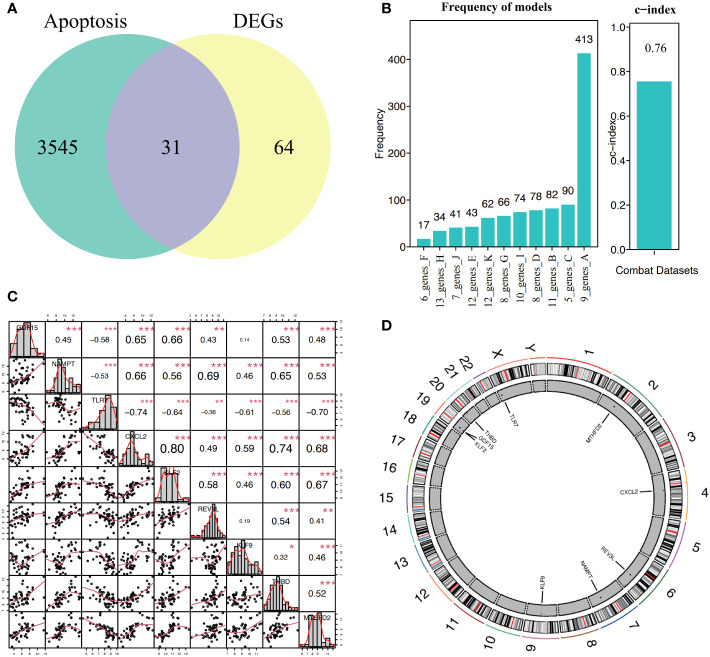
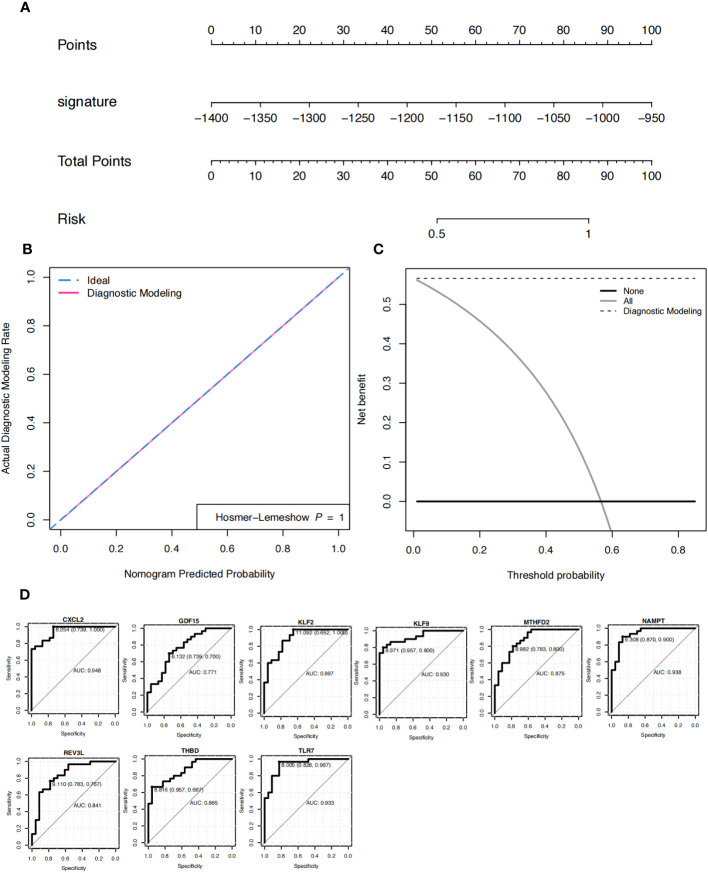
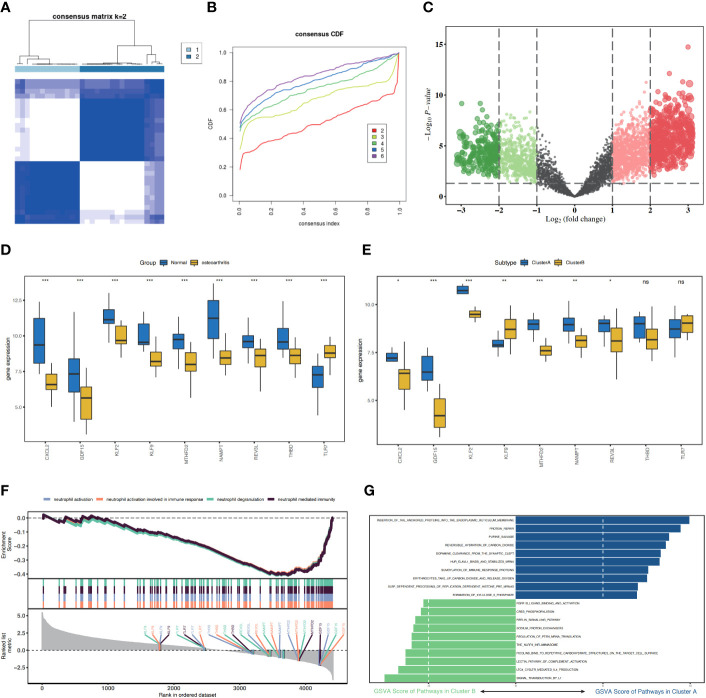
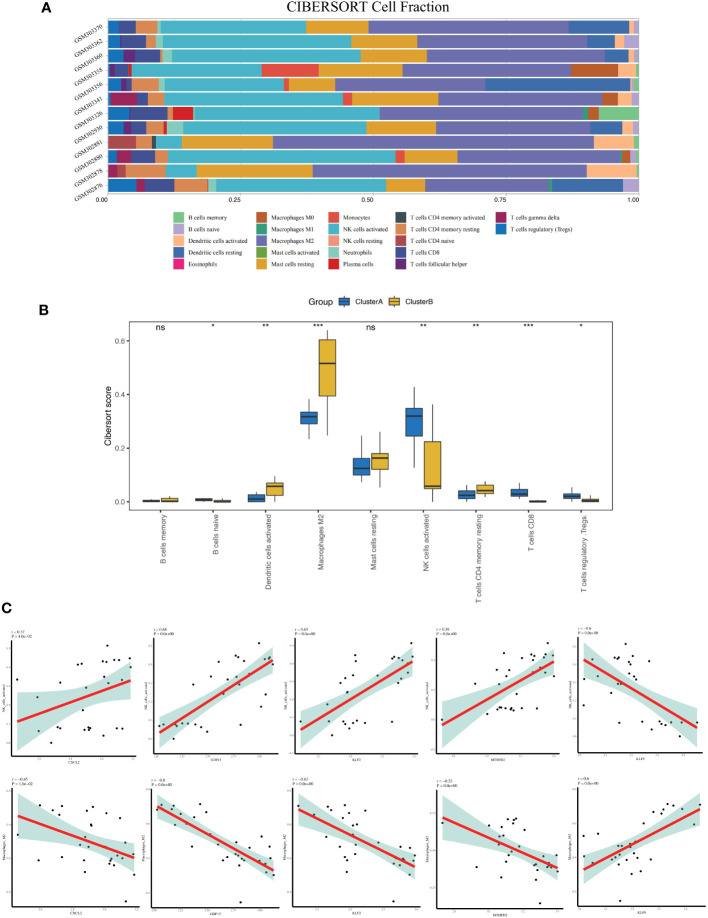
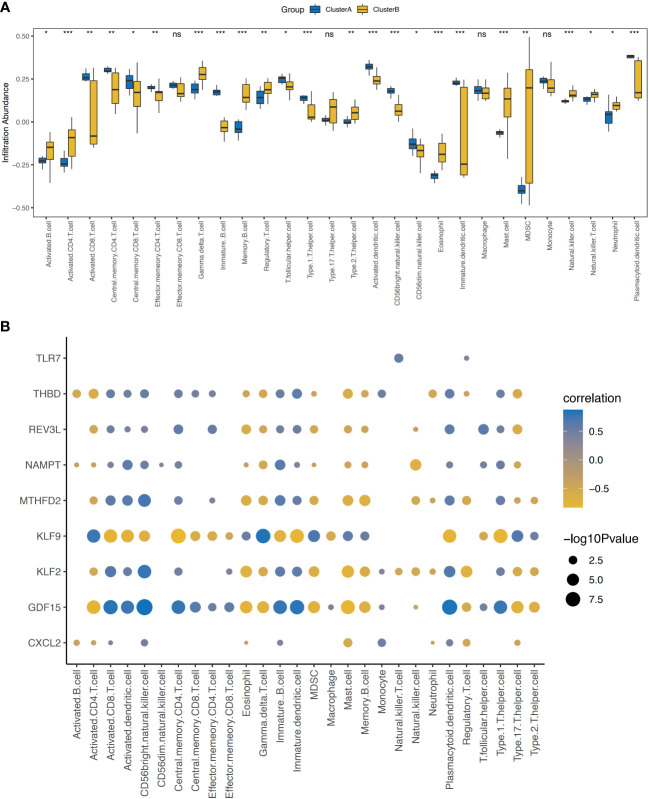
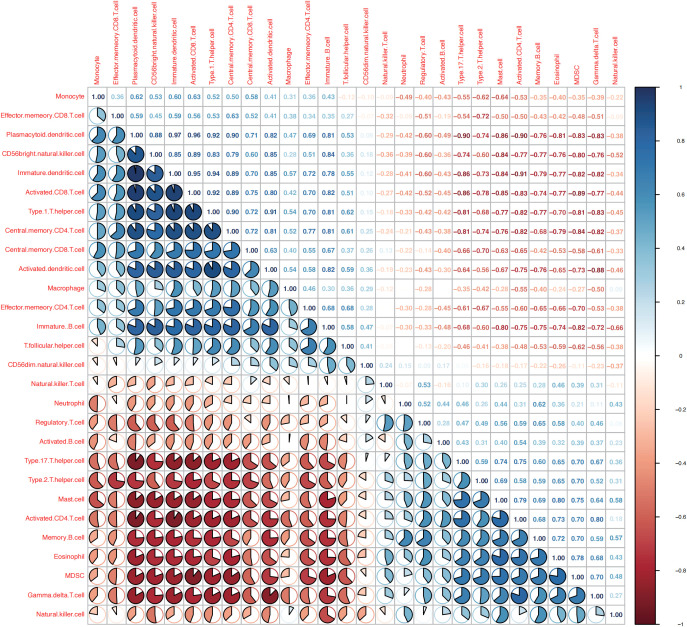
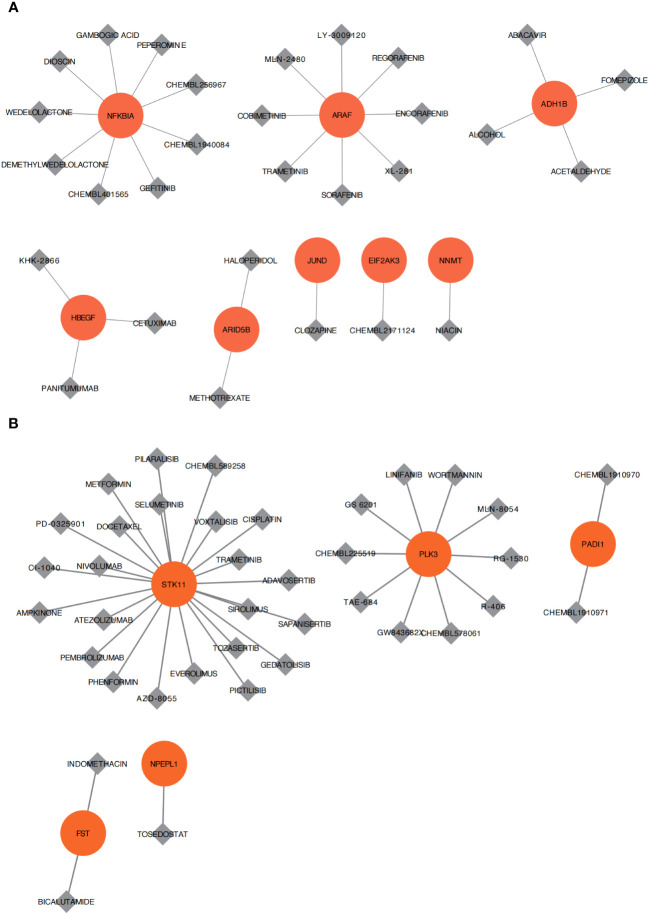
A total of 95 DEGs were identified, of which 47 were upregulated and 48 were downregulated, and 31 hub genes were selected as ARDEGs. LASSO regression analysis revealed nine characteristic genes: growth differentiation factor 15 (, , , , , , , , and . Clusters A and B were identified, and and were highly enriched in Cluster B, whereas and p signal pathways were enriched in Cluster A. The number of activated natural killer cells in Cluster B was significantly higher than that in Cluster A. and interacted with 193 and 32 TFs, respectively, and and interacted with 48 and 82 miRNAs, respectively.
ARGs could predict the occurrence of OA and may be related to different degrees of OA progression.
骨关节炎(OA)的进展涉及多种因素,包括作为退变基本病理机制的软骨侵蚀,并且与软骨细胞凋亡密切相关。为了分析凋亡与 OA 发展之间的相关性,我们从基因表达综合数据库(GEO)中 OA 和正常样本的差异表达基因(DEG)中选择凋亡基因,使用套索回归分析识别特征基因,并进行共识聚类分析以进一步探讨该疾病的发病机制。
从 GEO 下载 OA 样本的基因表达谱数据集 GSE12021 和 GSE55235,并进行组合分析以获得 DEG。从 GeneCards 数据库中收集凋亡相关基因(ARGs),并与 DEG 进行交集以获得凋亡相关 DEG(ARDEG)。使用最小绝对收缩和选择算子(LASSO)回归分析获得特征基因,并基于这些基因构建列线图。进行共识聚类分析将患者分为聚类。比较聚类的免疫特征、功能富集和免疫浸润状态。此外,构建了 mRNA 药物、mRNA-TF 和 mRNA-miRNA 的蛋白质-蛋白质相互作用网络。
共鉴定出 95 个 DEG,其中 47 个上调,48 个下调,选择 31 个 hub 基因作为 ARDEG。LASSO 回归分析揭示了 9 个特征基因:生长分化因子 15(、、、、、、、和)。鉴定出聚类 A 和聚类 B,和在聚类 B 中高度富集,而和 p 信号通路在聚类 A 中富集。聚类 B 中激活的自然杀伤细胞数量明显高于聚类 A。和分别与 193 和 32 个 TF 相互作用,和分别与 48 和 82 个 miRNA 相互作用。
ARGs 可以预测 OA 的发生,并且可能与 OA 进展的不同程度有关。