Lester and Sue Smith Breast Center, Baylor College of Medicine, Houston, Texas, USA
Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas, USA.
J Immunother Cancer. 2023 Oct;11(10). doi: 10.1136/jitc-2023-007073.
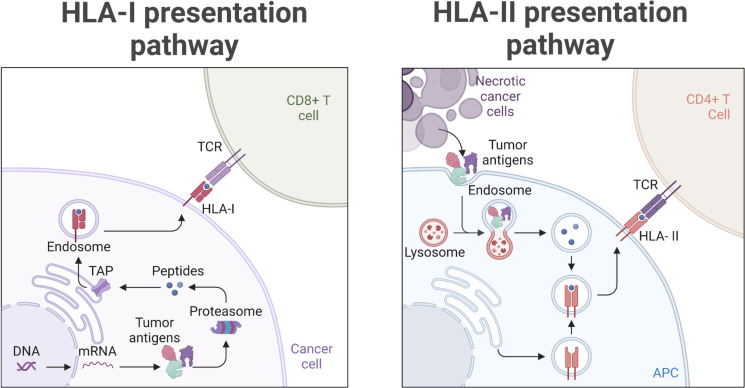
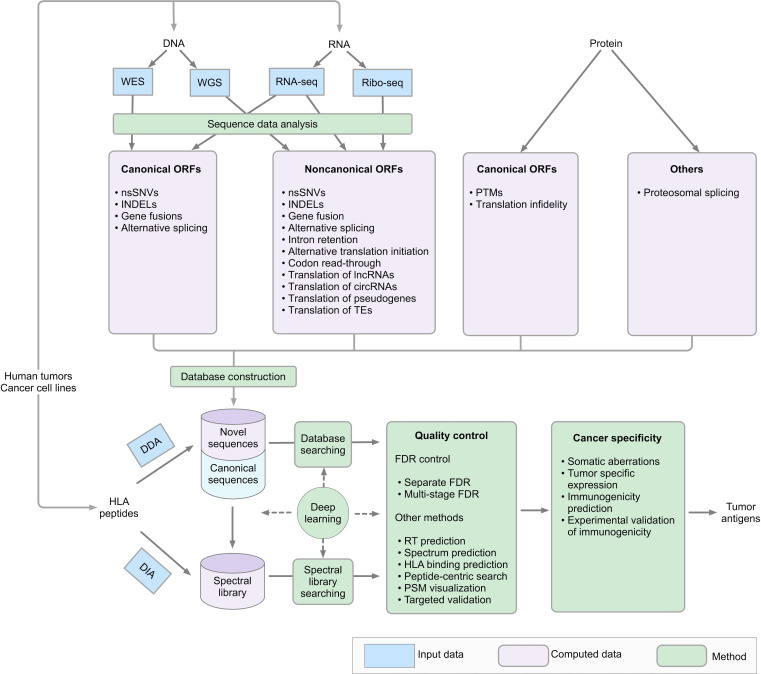
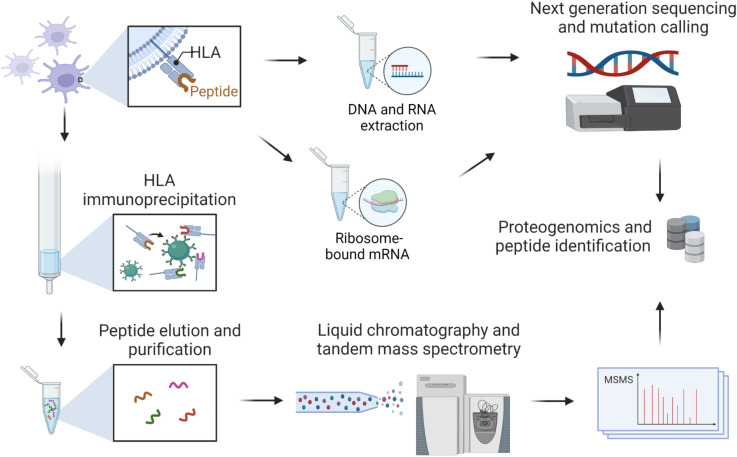
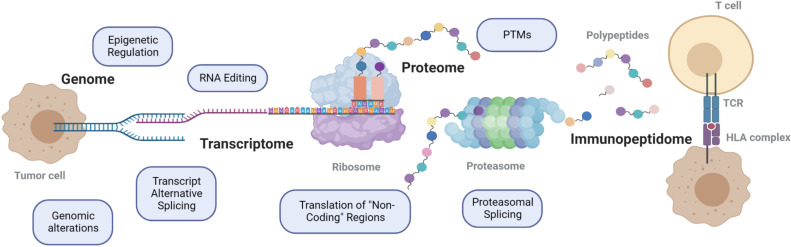
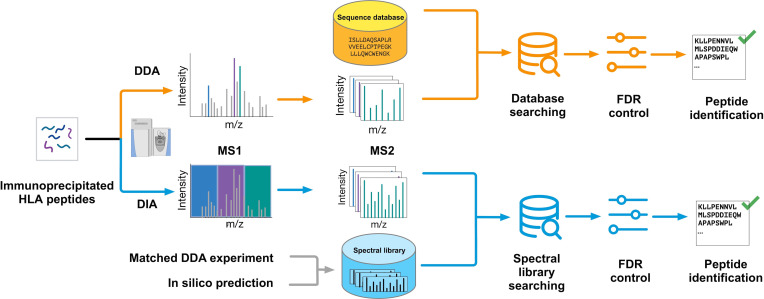
Identification of tumor antigens presented by the human leucocyte antigen (HLA) molecules is essential for the design of effective and safe cancer immunotherapies that rely on T cell recognition and killing of tumor cells. Mass spectrometry (MS)-based immunopeptidomics enables high-throughput, direct identification of HLA-bound peptides from a variety of cell lines, tumor tissues, and healthy tissues. It involves immunoaffinity purification of HLA complexes followed by MS profiling of the extracted peptides using data-dependent acquisition, data-independent acquisition, or targeted approaches. By incorporating DNA, RNA, and ribosome sequencing data into immunopeptidomics data analysis, the proteogenomic approach provides a powerful means for identifying tumor antigens encoded within the canonical open reading frames of annotated coding genes and non-canonical tumor antigens derived from presumably non-coding regions of our genome. We discuss emerging computational challenges in immunopeptidomics data analysis and tumor antigen identification, highlighting key considerations in the proteogenomics-based approach, including accurate DNA, RNA and ribosomal sequencing data analysis, careful incorporation of predicted novel protein sequences into reference protein database, special quality control in MS data analysis due to the expanded and heterogeneous search space, cancer-specificity determination, and immunogenicity prediction. The advancements in technology and computation is continually enabling us to identify tumor antigens with higher sensitivity and accuracy, paving the way toward the development of more effective cancer immunotherapies.
鉴定人类白细胞抗原(HLA)分子呈递的肿瘤抗原对于设计依赖 T 细胞识别和杀伤肿瘤细胞的有效和安全的癌症免疫疗法至关重要。基于质谱(MS)的免疫肽组学能够从各种细胞系、肿瘤组织和健康组织中高通量、直接鉴定与 HLA 结合的肽。它涉及 HLA 复合物的免疫亲和纯化,然后使用依赖数据的采集、独立数据的采集或靶向方法对提取的肽进行 MS 分析。通过将 DNA、RNA 和核糖体测序数据纳入免疫肽组学数据分析中,蛋白质基因组学方法为鉴定编码在注释编码基因的典型开放阅读框内的肿瘤抗原以及来自我们基因组的可能非编码区域的非典型肿瘤抗原提供了一种强大的手段。我们讨论了免疫肽组学数据分析和肿瘤抗原鉴定中出现的计算挑战,强调了基于蛋白质基因组学方法的关键考虑因素,包括准确的 DNA、RNA 和核糖体测序数据分析、仔细将预测的新蛋白序列纳入参考蛋白数据库、由于扩展和异质搜索空间,在 MS 数据分析中进行特殊的质量控制、癌症特异性确定和免疫原性预测。技术和计算的进步不断使我们能够以更高的灵敏度和准确性识别肿瘤抗原,为开发更有效的癌症免疫疗法铺平道路。