Kosmopoulos James C, Klier Katherine M, Langwig Marguerite V, Tran Patricia Q, Anantharaman Karthik
Department of Bacteriology, University of Wisconsin-Madison, Madison, Wisconsin, USA.
Microbiology Doctoral Training Program, University of Wisconsin-Madison, Madison, Wisconsin, USA.
bioRxiv. 2023 Dec 12:2023.10.15.562385. doi: 10.1101/2023.10.15.562385.
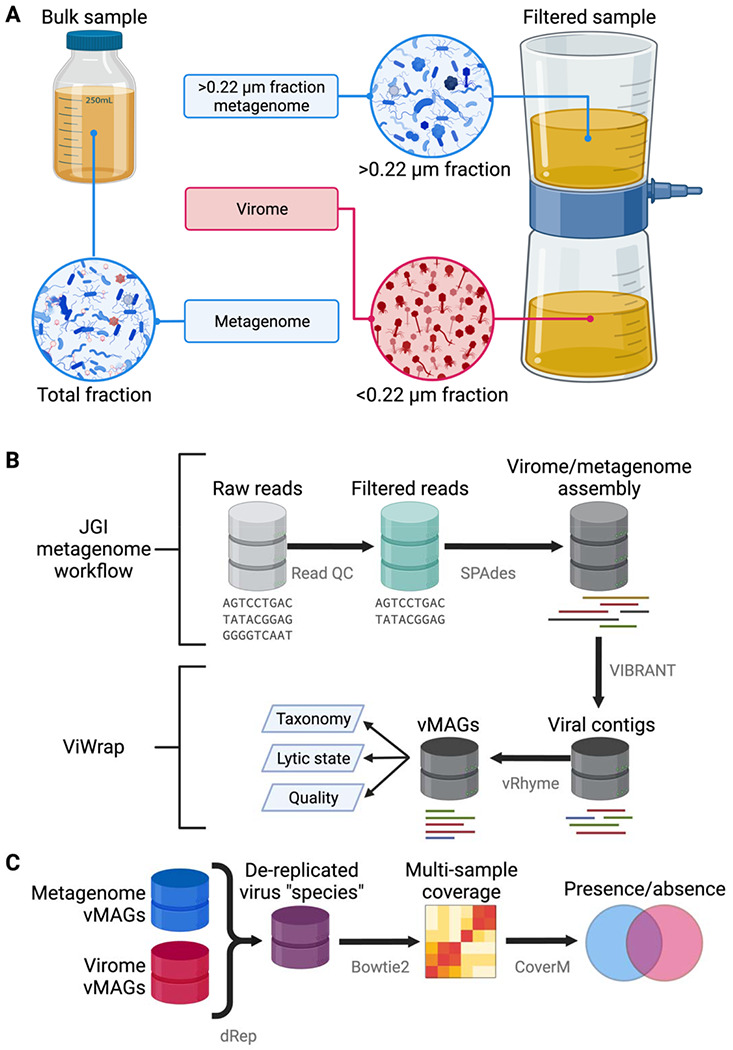
Viruses, the majority of which are uncultivated, are among the most abundant biological entities on Earth. From altering microbial physiology to driving community dynamics, viruses are fundamental members of microbiomes. While the number of studies leveraging viral metagenomics (viromics) for studying uncultivated viruses is growing, standards for viromics research are lacking. Viromics can utilize computational discovery of viruses from total metagenomes of all community members (hereafter metagenomes) or use physical separation of virus-specific fractions (hereafter viromes). However, differences in the recovery and interpretation of viruses from metagenomes and viromes obtained from the same samples remain understudied.
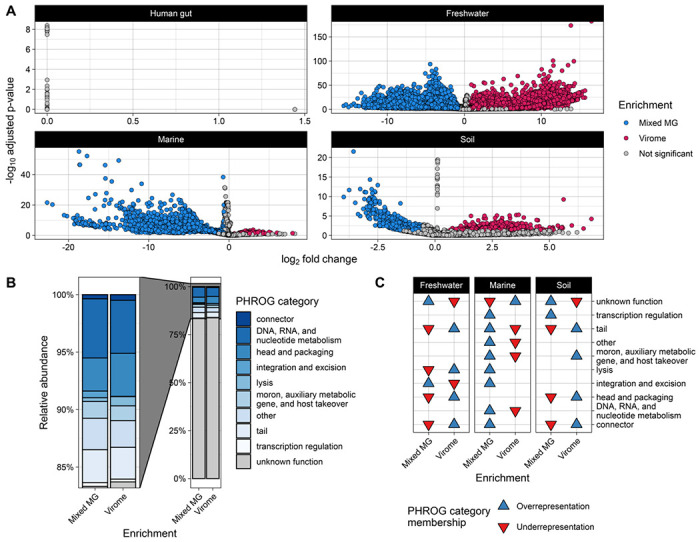
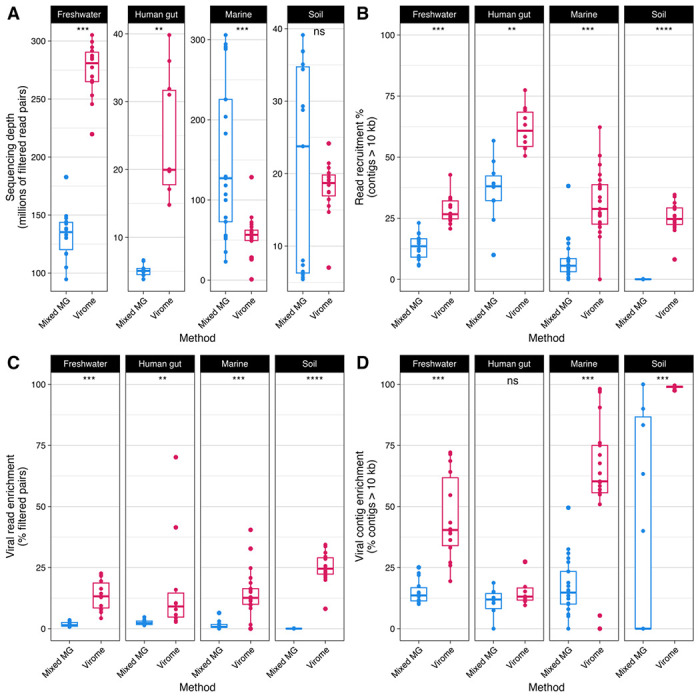
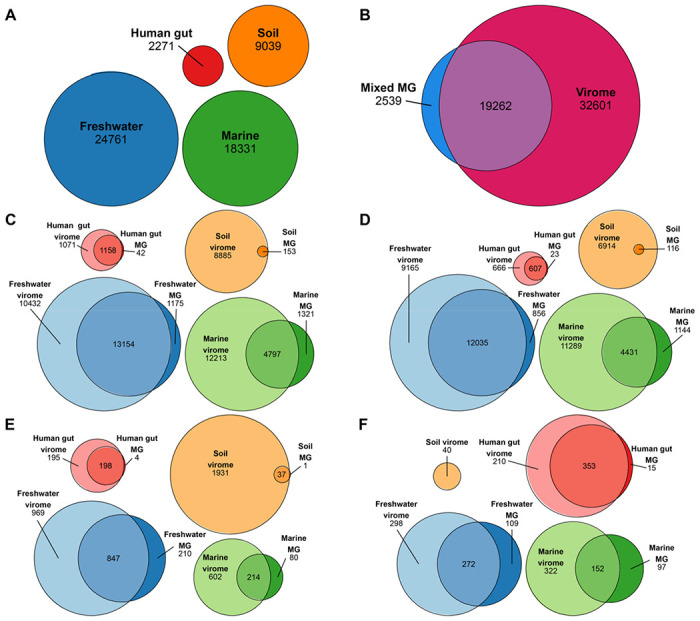
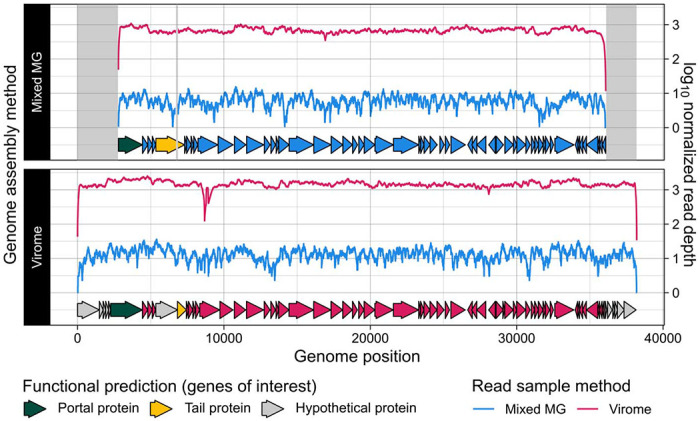
Here, we compare viral communities from paired viromes and metagenomes obtained from 60 diverse samples across human gut, soil, freshwater, and marine ecosystems. Overall, viral communities obtained from viromes were more abundant and species rich than those obtained from metagenomes, although there were some exceptions. Despite this, metagenomes still contained many viral genomes not detected in viromes. We also found notable differences in the predicted lytic state of viruses detected in viromes vs metagenomes at the time of sequencing. Other forms of variation observed include genome presence/absence, genome quality, and encoded protein content between viromes and metagenomes, but the magnitude of these differences varied by environment.
Overall, our results show that the choice of method can lead to differing interpretations of viral community ecology. We suggest that the choice of whether to target a metagenome or virome to study viral communities should be dependent on the environmental context and ecological questions being asked. However, our overall recommendation to researchers investigating viral ecology and evolution is to pair both approaches to maximize their respective benefits.
病毒是地球上数量最为丰富的生物实体之一,其中大部分尚未被培养。从改变微生物生理机能到推动群落动态变化,病毒都是微生物群落的重要组成部分。虽然利用病毒宏基因组学(病毒组学)研究未培养病毒的研究数量不断增加,但病毒组学研究的标准仍不完善。病毒组学可以通过计算从所有群落成员的总宏基因组(以下简称宏基因组)中发现病毒,也可以对病毒特异性组分进行物理分离(以下简称病毒组)。然而,从相同样本中获得的宏基因组和病毒组在病毒回收和解读方面的差异仍未得到充分研究。
在这里,我们比较了从人类肠道、土壤、淡水和海洋生态系统的60个不同样本中获得的配对病毒组和宏基因组中的病毒群落。总体而言,从病毒组中获得的病毒群落比从宏基因组中获得的更为丰富且物种更多,不过也有一些例外情况。尽管如此,宏基因组中仍包含许多在病毒组中未检测到的病毒基因组。我们还发现在测序时,病毒组和宏基因组中检测到的病毒预测裂解状态存在显著差异。观察到其他形式的变异包括病毒组和宏基因组之间基因组的有无、基因组质量以及编码蛋白含量,但这些差异的程度因环境而异。
总体而言,我们的结果表明方法的选择会导致对病毒群落生态学的不同解读。我们建议,选择针对宏基因组还是病毒组来研究病毒群落应取决于环境背景和所提出的生态问题。然而,我们对研究病毒生态学和进化的研究人员的总体建议是将两种方法结合使用,以最大限度地发挥各自的优势。