SCI Population Biobanking & Translational Research Group, Swiss Paraplegic Research, Nottwil, Switzerland.
Department of Orthopaedic Surgery, University of Bern, Bern Inselspital, Bern, Switzerland.
BMC Microbiol. 2024 Feb 16;24(1):58. doi: 10.1186/s12866-024-03208-5.
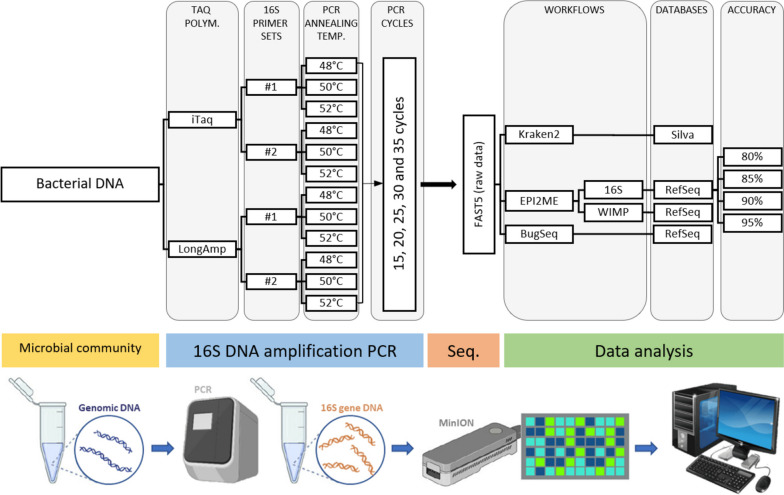
Accurate identification of bacterial communities is crucial for research applications, diagnostics, and clinical interventions. Although 16S ribosomal RNA (rRNA) gene sequencing is a widely employed technique for bacterial taxonomic classification, it often results in misclassified or unclassified bacterial taxa. This study sought to refine the full-length 16S rRNA gene sequencing protocol using the MinION sequencer, focusing on the V1-V9 regions. Our methodological enquiry examined several factors, including the number of PCR amplification cycles, choice of primers and Taq polymerase, and specific sequence databases and workflows employed. We used a microbial standard comprising eight bacterial strains (five gram-positive and three gram-negative) in known proportions as a validation control.
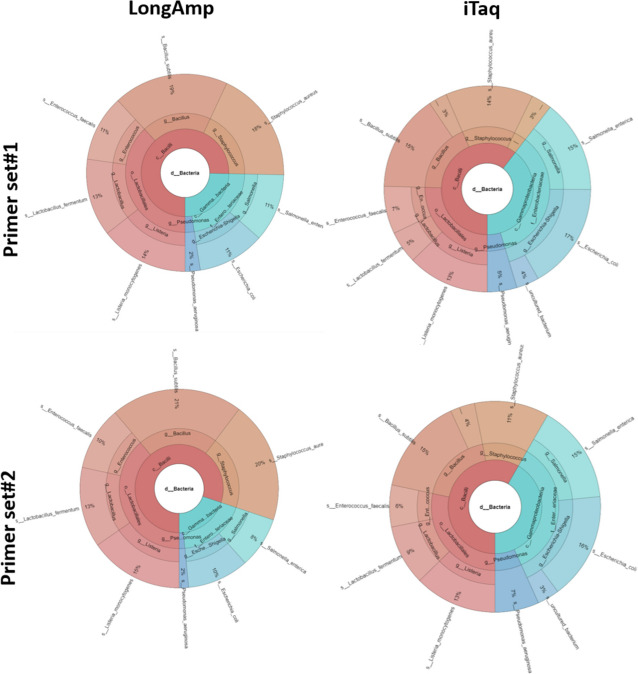
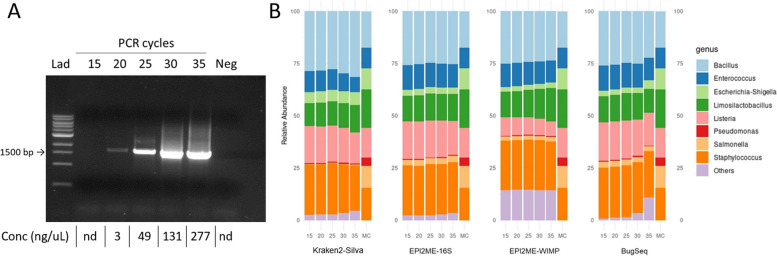
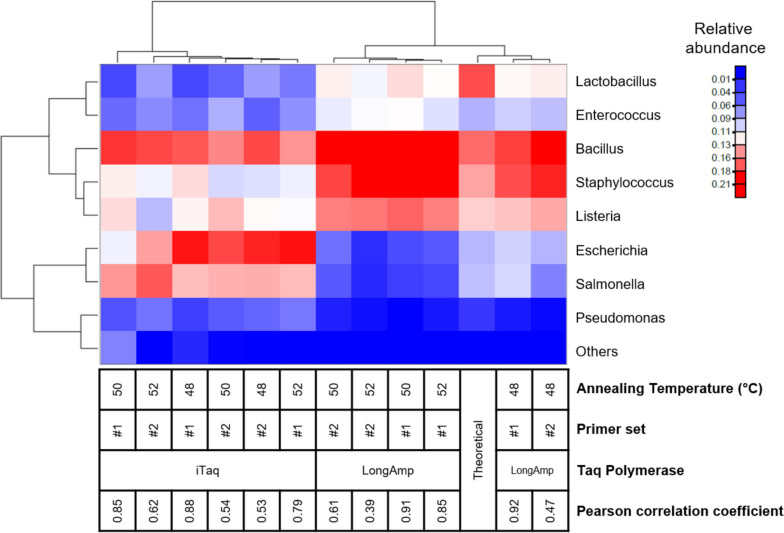
Based on the MinION protocol, we employed the microbial standard as the DNA template for the 16S rRNA gene amplicon sequencing procedure. Our analysis showed that an elevated number of PCR amplification cycles introduced PCR bias, and the selection of Taq polymerase and primer sets significantly affected the subsequent analysis. Bacterial identification at genus level demonstrated Pearson correlation coefficients ranging from 0.73 to 0.79 when assessed using BugSeq, Kraken-Silva and EPI2ME-16S workflows. Notably, the EPI2ME-16S workflow exhibited the highest Pearson correlation with the microbial standard, minimised misclassification, and increased alignment accuracy. At the species taxonomic level, the BugSeq workflow was superior, with a Pearson correlation coefficient of 0.92.
These findings emphasise the importance of careful selection of PCR settings and a well-structured analytical framework for 16S rRNA full-length gene sequencing. The results showed a robust correlation between the predicted and observed bacterial abundances at both the genus and species taxonomic levels, making these findings applicable across diverse research contexts and with clinical utility for reliable pathogen identification.
准确识别细菌群落对于研究应用、诊断和临床干预至关重要。尽管 16S 核糖体 RNA(rRNA)基因测序是一种广泛用于细菌分类学的技术,但它通常会导致细菌分类单元的错误分类或未分类。本研究旨在使用 MinION 测序仪优化全长 16S rRNA 基因测序方案,重点关注 V1-V9 区。我们的方法研究考察了几个因素,包括 PCR 扩增循环次数、引物和 Taq 聚合酶的选择,以及使用的特定序列数据库和工作流程。我们使用包含已知比例的八种细菌菌株(五种革兰氏阳性和三种革兰氏阴性)的微生物标准作为验证对照。
根据 MinION 方案,我们将微生物标准用作 16S rRNA 基因扩增子测序程序的 DNA 模板。我们的分析表明,增加 PCR 扩增循环次数会引入 PCR 偏差,并且 Taq 聚合酶和引物组的选择会显著影响后续分析。使用 BugSeq、Kraken-Silva 和 EPI2ME-16S 工作流程评估时,细菌在属水平的鉴定显示 Pearson 相关系数在 0.73 到 0.79 之间。值得注意的是,EPI2ME-16S 工作流程与微生物标准的 Pearson 相关系数最高,最小化了错误分类并提高了对齐准确性。在种分类水平上,BugSeq 工作流程表现最佳,Pearson 相关系数为 0.92。
这些发现强调了在进行全长 16S rRNA 基因测序时仔细选择 PCR 设置和构建良好的分析框架的重要性。结果表明,在属和种分类水平上,预测和观察到的细菌丰度之间存在稳健的相关性,这使得这些发现适用于各种研究背景,并具有可靠的病原体鉴定的临床应用价值。