Section of Cardio and Respiratory Medicine, Department of Medicine, Victor Phillip Dahdaleh Heart and Lung Research Institute, Papworth Road, Cambridge CB2 0BB, UK.
Department of Cellular and Molecular Medicine, KU Leuven, Herestraat 49, Box 802, 3000 Leuven, Belgium.
Cardiovasc Res. 2024 May 29;120(7):756-768. doi: 10.1093/cvr/cvae068.
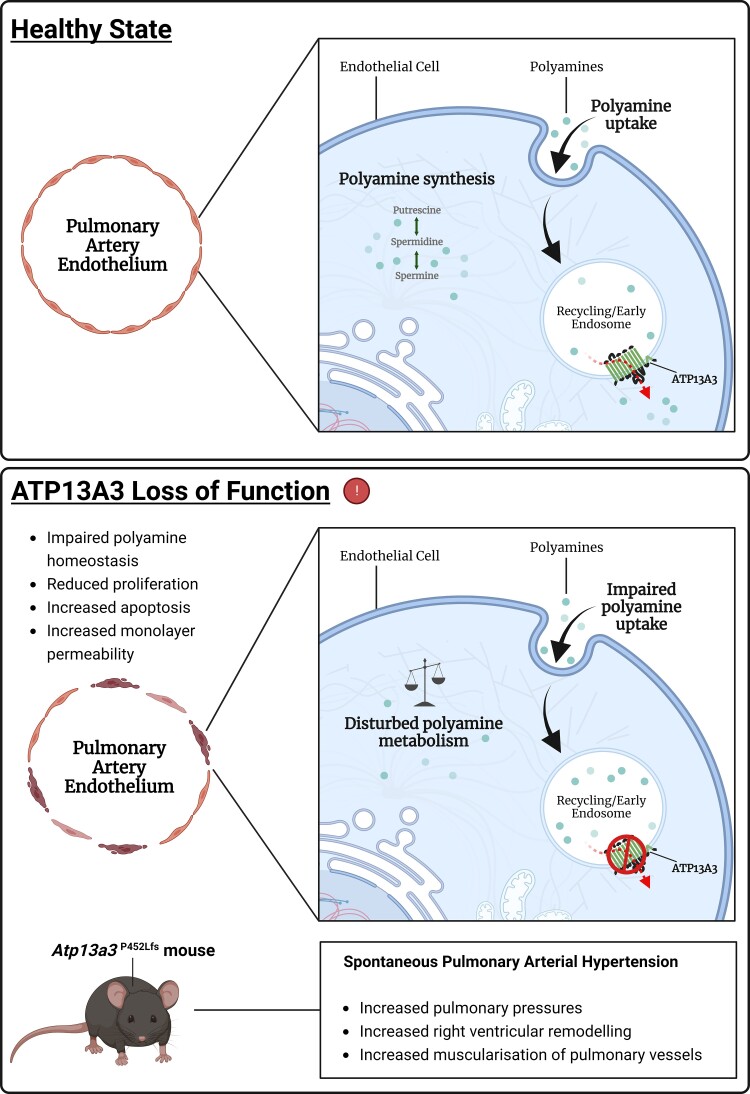
Potential loss-of-function variants of ATP13A3, the gene encoding a P5B-type transport ATPase of undefined function, were recently identified in patients with pulmonary arterial hypertension (PAH). ATP13A3 is implicated in polyamine transport but its function has not been fully elucidated. In this study, we sought to determine the biological function of ATP13A3 in vascular endothelial cells (ECs) and how PAH-associated variants may contribute to disease pathogenesis.
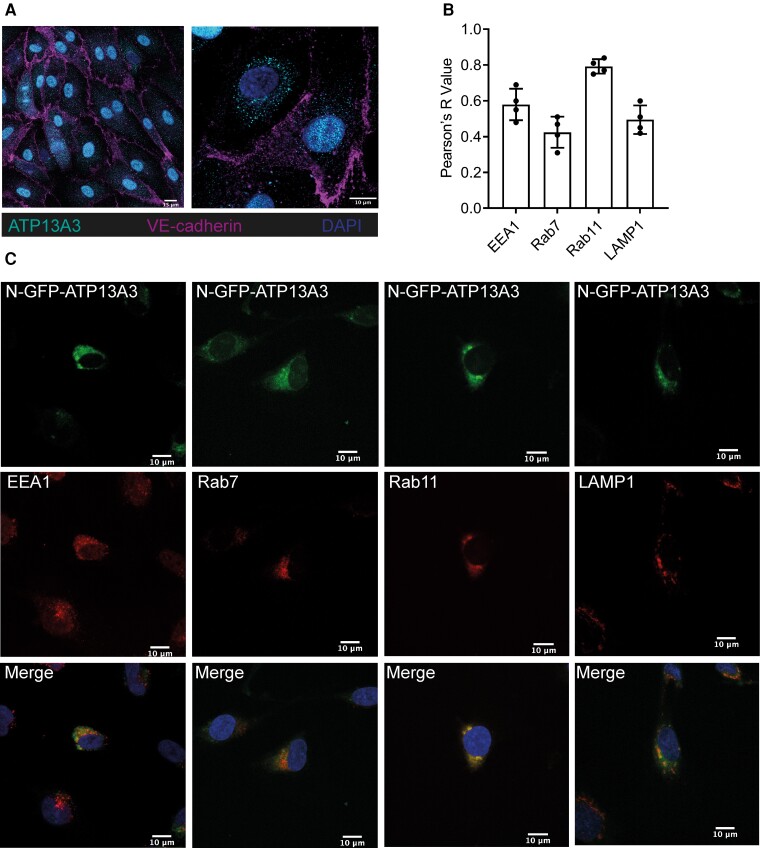
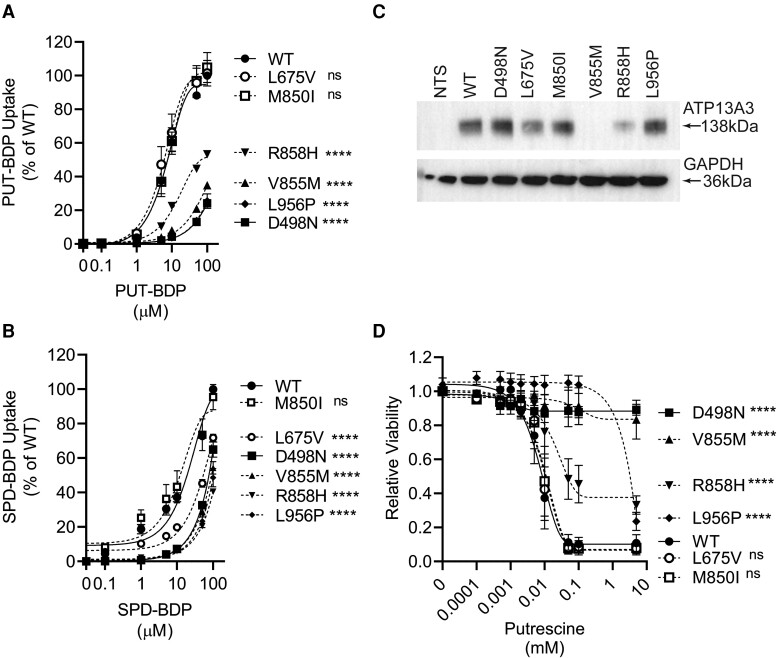
We studied the impact of ATP13A3 deficiency and overexpression in EC models [human pulmonary ECs, blood outgrowth ECs (BOECs), and human microvascular EC 1], including a PAH patient-derived BOEC line harbouring an ATP13A3 variant (LK726X). We also generated mice harbouring an Atp13a3 variant analogous to a human disease-associated variant to establish whether these mice develop PAH. ATP13A3 localized to the recycling endosomes of human ECs. Knockdown of ATP13A3 in ECs generally reduced the basal polyamine content and altered the expression of enzymes involved in polyamine metabolism. Conversely, overexpression of wild-type ATP13A3 increased polyamine uptake. Functionally, loss of ATP13A3 was associated with reduced EC proliferation, increased apoptosis in serum starvation, and increased monolayer permeability to thrombin. The assessment of five PAH-associated missense ATP13A3 variants (L675V, M850I, V855M, R858H, and L956P) confirmed loss-of-function phenotypes represented by impaired polyamine transport and dysregulated EC function. Furthermore, mice carrying a heterozygous germline Atp13a3 frameshift variant representing a human variant spontaneously developed a PAH phenotype, with increased pulmonary pressures, right ventricular remodelling, and muscularization of pulmonary vessels.
We identify ATP13A3 as a polyamine transporter controlling polyamine homeostasis in ECs, a deficiency of which leads to EC dysfunction and predisposes to PAH. This suggests a need for targeted therapies to alleviate the imbalances in polyamine homeostasis and EC dysfunction in PAH.
最近在肺动脉高压(PAH)患者中发现了 ATP13A3 基因(编码一种功能未知的 P5B 型转运 ATP 酶)的潜在功能丧失变异体。ATP13A3 参与多胺转运,但功能尚未完全阐明。在这项研究中,我们试图确定 ATP13A3 在血管内皮细胞(ECs)中的生物学功能,以及与 PAH 相关的变异体如何导致疾病发病机制。
我们研究了 EC 模型(人肺 ECs、血液外生 ECs(BOECs)和人微血管 EC1)中 ATP13A3 缺乏和过表达的影响,包括携带 ATP13A3 变异体(LK726X)的 PAH 患者来源的 BOEC 系。我们还生成了携带类似于人类疾病相关变异体的 Atp13a3 变异体的小鼠,以确定这些小鼠是否会发展为 PAH。ATP13A3 定位于人 EC 的再循环内体。在 EC 中敲低 ATP13A3 通常会降低基础多胺含量并改变多胺代谢相关酶的表达。相反,野生型 ATP13A3 的过表达会增加多胺摄取。功能上,ATP13A3 的缺失与 EC 增殖减少、血清饥饿时凋亡增加以及凝血酶单层通透性增加有关。对五个与 PAH 相关的错义 ATP13A3 变异体(L675V、M850I、V855M、R858H 和 L956P)的评估证实了多胺转运受损和 EC 功能失调的功能丧失表型。此外,携带代表人类变异体的杂合性种系 Atp13a3 移码变异体的小鼠自发地发展出 PAH 表型,表现为肺动脉压升高、右心室重构和肺血管肌化。
我们将 ATP13A3 鉴定为一种多胺转运体,可控制 EC 中的多胺动态平衡,其缺乏会导致 EC 功能障碍并易患 PAH。这表明需要针对多胺动态平衡和 PAH 中 EC 功能障碍的靶向治疗。