Department of Biohealth Informatics, Luddy School of Informatics, Computing, and Engineering, Indiana University Indianapolis (IUI), Indianapolis, Indiana, USA.
Computers and Systems Department, National Telecommunication Institute, Cairo, Egypt.
RNA Biol. 2024 Jan;21(1):1-15. doi: 10.1080/15476286.2024.2352192. Epub 2024 May 17.
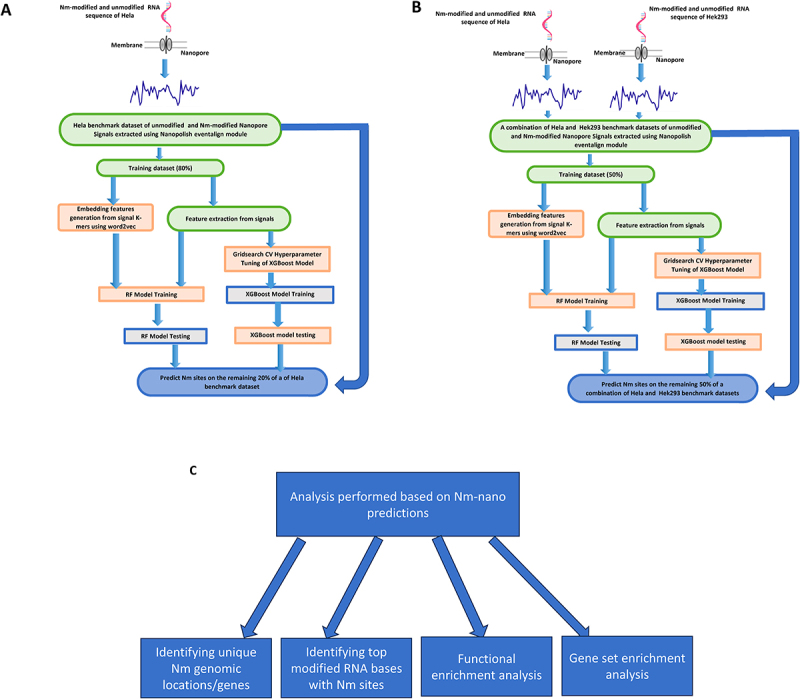
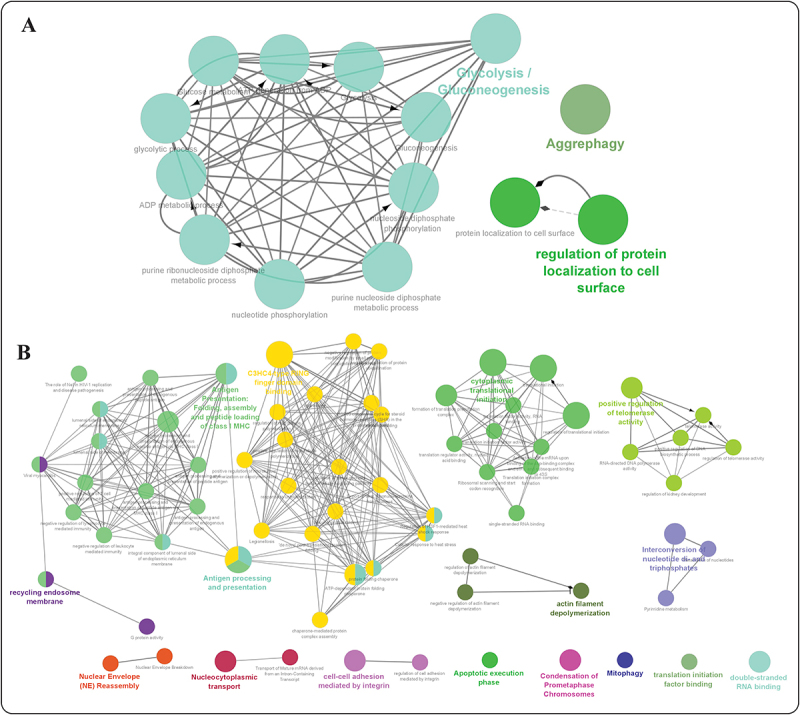
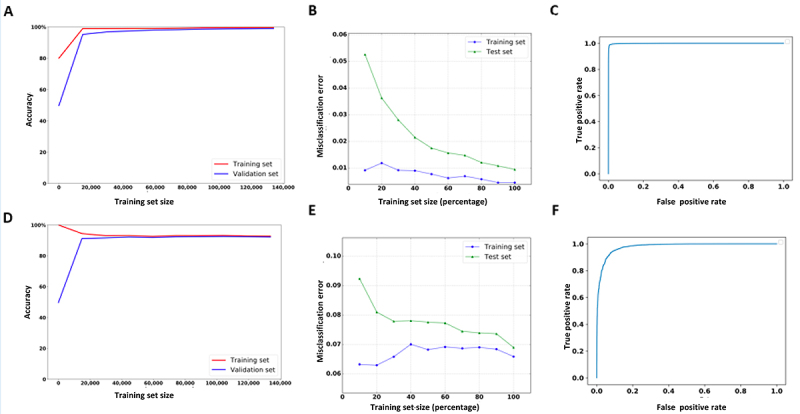
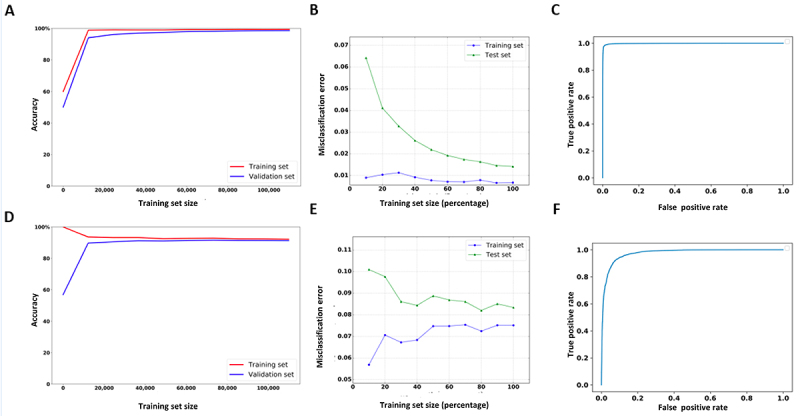
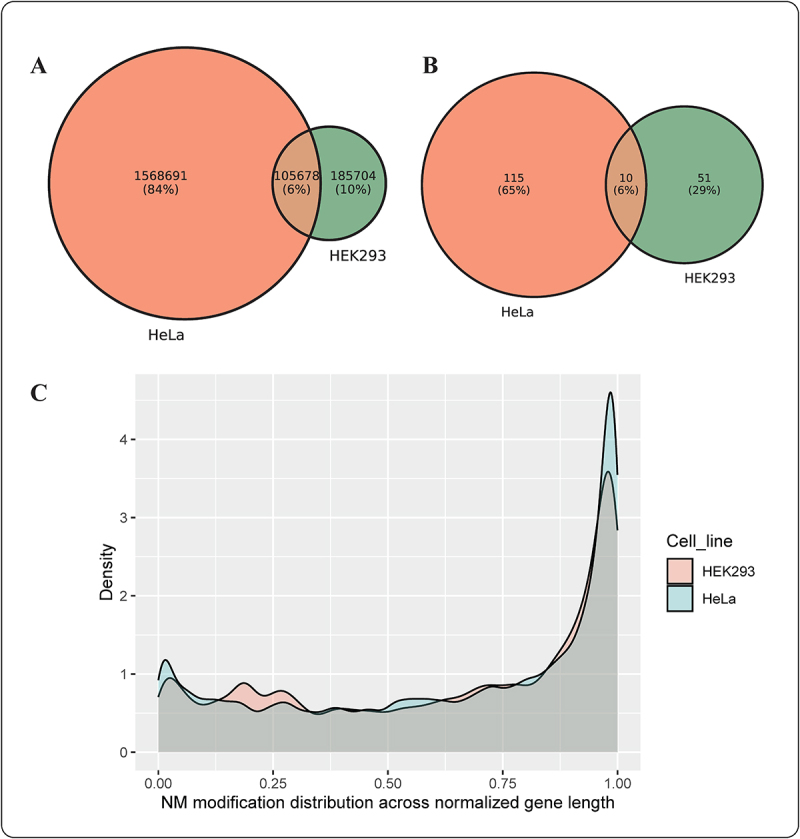
2´-O-methylation (Nm) is one of the most abundant modifications found in both mRNAs and noncoding RNAs. It contributes to many biological processes, such as the normal functioning of tRNA, the protection of mRNA against degradation by the decapping and exoribonuclease (DXO) protein, and the biogenesis and specificity of rRNA. Recent advancements in single-molecule sequencing techniques for long read RNA sequencing data offered by Oxford Nanopore technologies have enabled the direct detection of RNA modifications from sequencing data. In this study, we propose a bio-computational framework, Nm-Nano, for predicting the presence of Nm sites in direct RNA sequencing data generated from two human cell lines. The Nm-Nano framework integrates two supervised machine learning (ML) models for predicting Nm sites: Extreme Gradient Boosting (XGBoost) and Random Forest (RF) with K-mer embedding. Evaluation on benchmark datasets from direct RNA sequecing of HeLa and HEK293 cell lines, demonstrates high accuracy (99% with XGBoost and 92% with RF) in identifying Nm sites. Deploying Nm-Nano on HeLa and HEK293 cell lines reveals genes that are frequently modified with Nm. In HeLa cell lines, 125 genes are identified as frequently Nm-modified, showing enrichment in 30 ontologies related to immune response and cellular processes. In HEK293 cell lines, 61 genes are identified as frequently Nm-modified, with enrichment in processes like glycolysis and protein localization. These findings underscore the diverse regulatory roles of Nm modifications in metabolic pathways, protein degradation, and cellular processes. The source code of Nm-Nano can be freely accessed at https://github.com/Janga-Lab/Nm-Nano.
2´-O-甲基化(Nm)是在 mRNA 和非编码 RNA 中发现的最丰富的修饰之一。它有助于许多生物过程,如 tRNA 的正常功能、mRNA 免受脱帽和外切核酸酶(DXO)蛋白降解的保护,以及 rRNA 的生物发生和特异性。牛津纳米孔技术提供的长读 RNA 测序数据的单分子测序技术的最新进展使得能够直接从测序数据中检测 RNA 修饰。在这项研究中,我们提出了一个生物计算框架 Nm-Nano,用于预测来自两种人类细胞系的直接 RNA 测序数据中 Nm 位点的存在。Nm-Nano 框架集成了两个用于预测 Nm 位点的监督机器学习(ML)模型:极端梯度提升(XGBoost)和随机森林(RF)与 K-mer 嵌入。在直接 RNA 测序的基准数据集上对 HeLa 和 HEK293 细胞系的评估表明,在识别 Nm 位点方面具有很高的准确性(XGBoost 为 99%,RF 为 92%)。在 HeLa 和 HEK293 细胞系上部署 Nm-Nano 揭示了经常被 Nm 修饰的基因。在 HeLa 细胞系中,鉴定出 125 个经常被 Nm 修饰的基因,这些基因富集在与免疫反应和细胞过程相关的 30 个本体中。在 HEK293 细胞系中,鉴定出 61 个经常被 Nm 修饰的基因,这些基因富集在糖酵解和蛋白质定位等过程中。这些发现强调了 Nm 修饰在代谢途径、蛋白质降解和细胞过程中的多样化调节作用。Nm-Nano 的源代码可在 https://github.com/Janga-Lab/Nm-Nano 上免费访问。