Department of Urology, the First Affiliated Hospital of Nanjing Medical University, Nanjing, China.
Department of Urology, the Second Affiliated Hospital of Nanjing Medical University, Nanjing, China.
Clin Transl Med. 2024 May;14(5):e1686. doi: 10.1002/ctm2.1686.
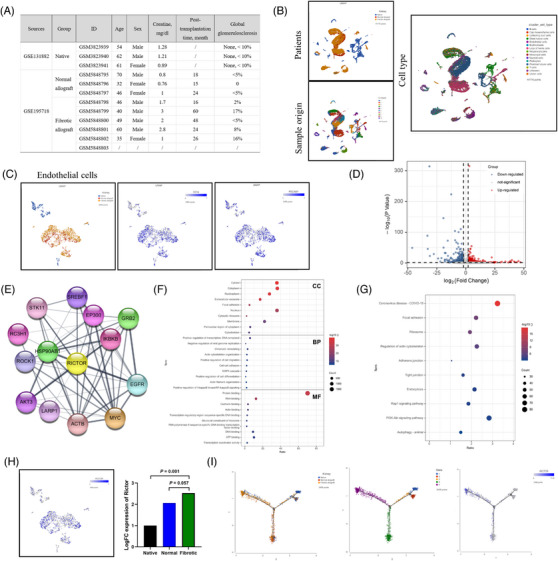
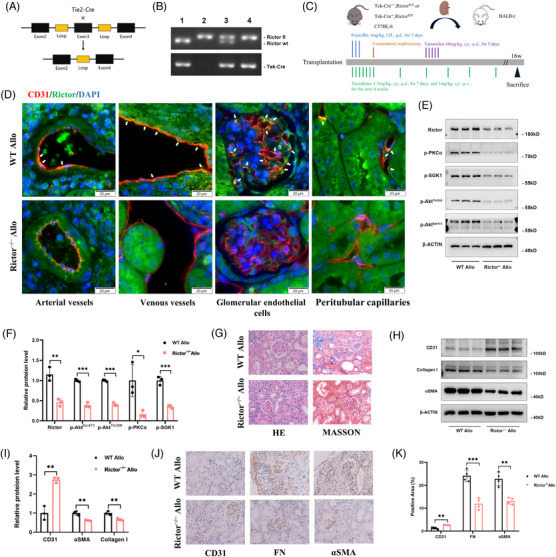
Renal allograft interstitial fibrosis/tubular atrophy (IF/TA) constitutes the principal histopathological characteristic of chronic allograft dysfunction (CAD) in kidney-transplanted patients. While renal vascular endothelial-mesenchymal transition (EndMT) has been verified as an important contributing factor to IF/TA in CAD patients, its underlying mechanisms remain obscure. Through single-cell transcriptomic analysis, we identified Rictor as a potential pivotal mediator for EndMT. This investigation sought to elucidate the role of Rictor/mTORC2 signalling in the pathogenesis of renal allograft interstitial fibrosis and the associated mechanisms.
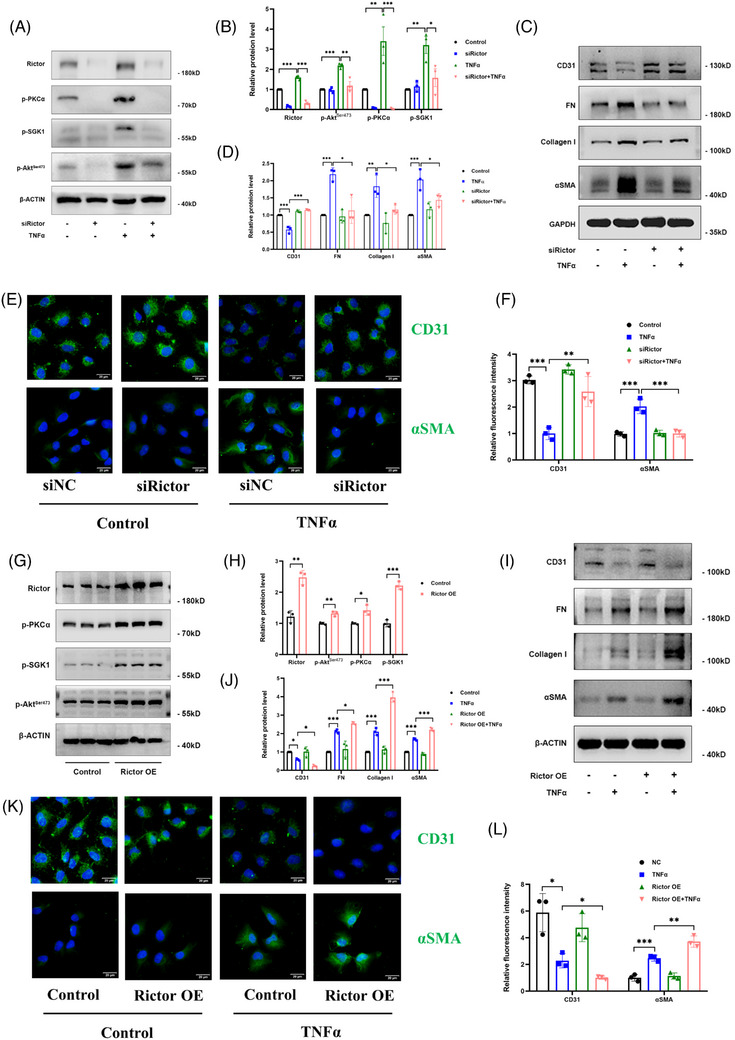
The influence of the Rictor/mTOR2 pathway on renal vascular EndMT and renal allograft fibrosis was investigated by cell experiments and Rictor depletion in renal allogeneic transplantation mice models. Subsequently, a series of assays were conducted to explore the underlying mechanisms of the enhanced mitophagy and the ameliorated EndMT resulting from Rictor knockout.
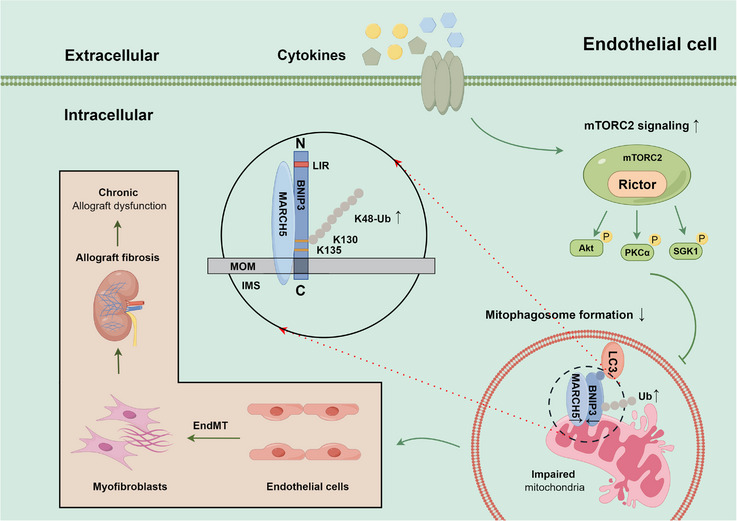
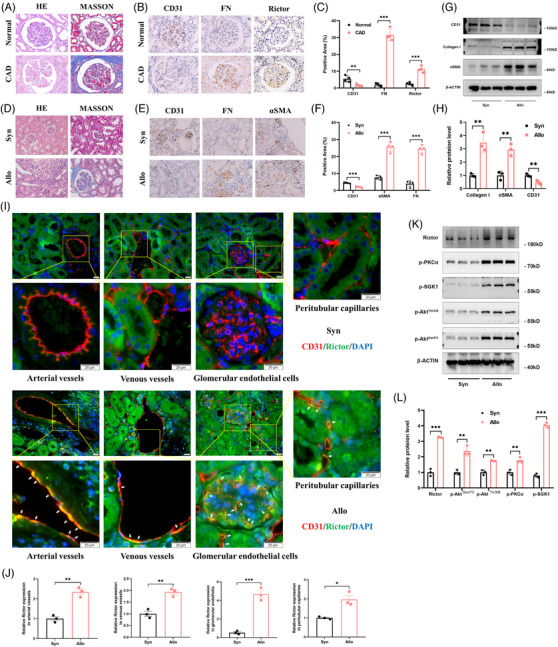
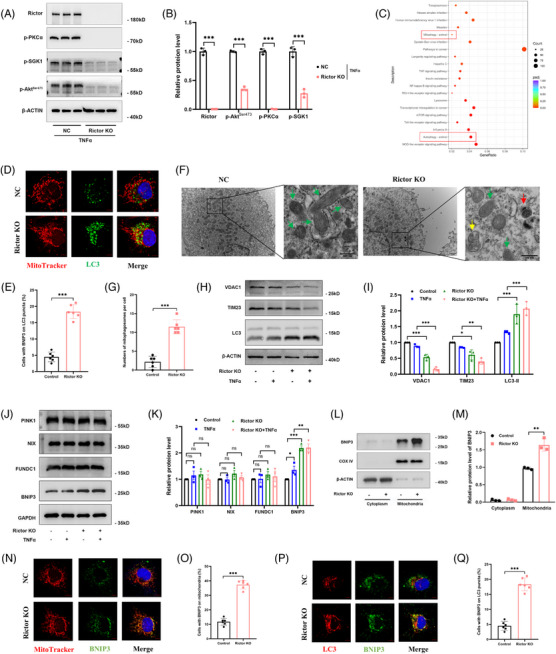
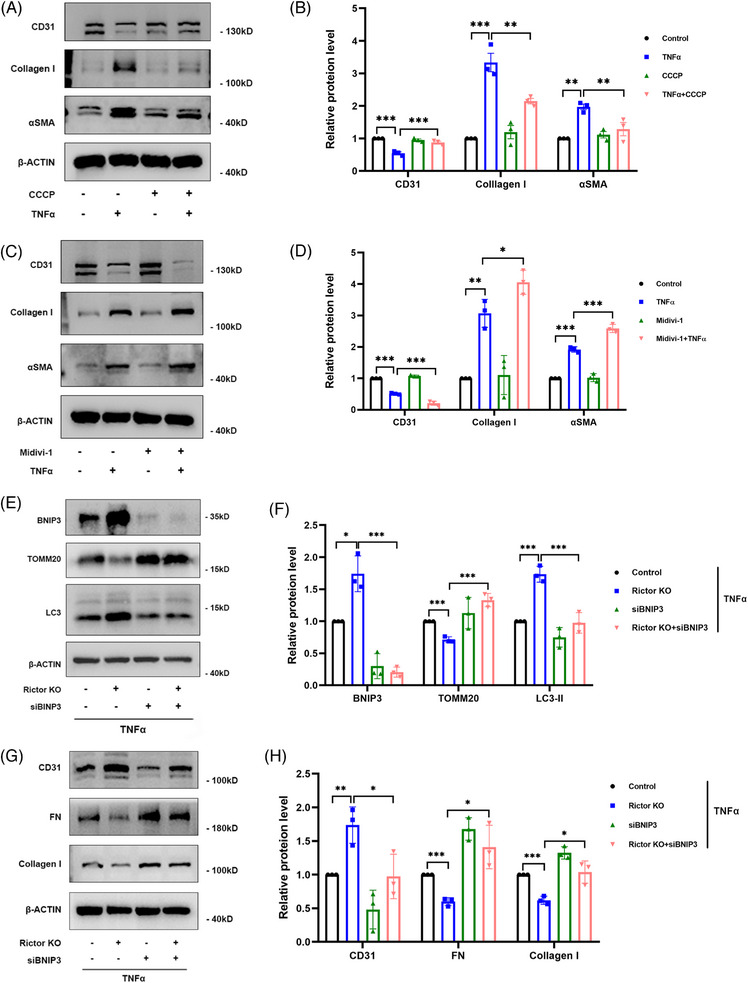
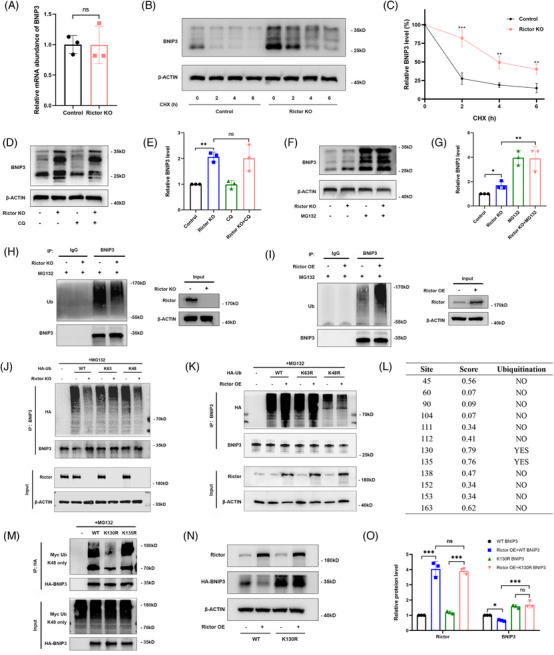
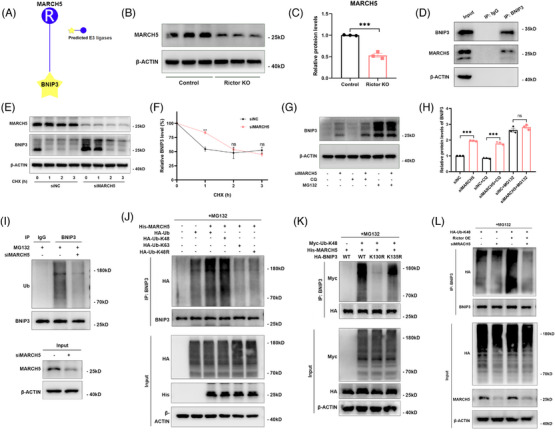
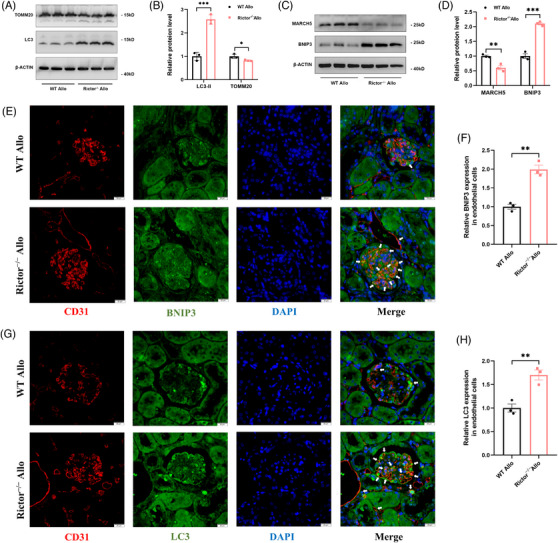
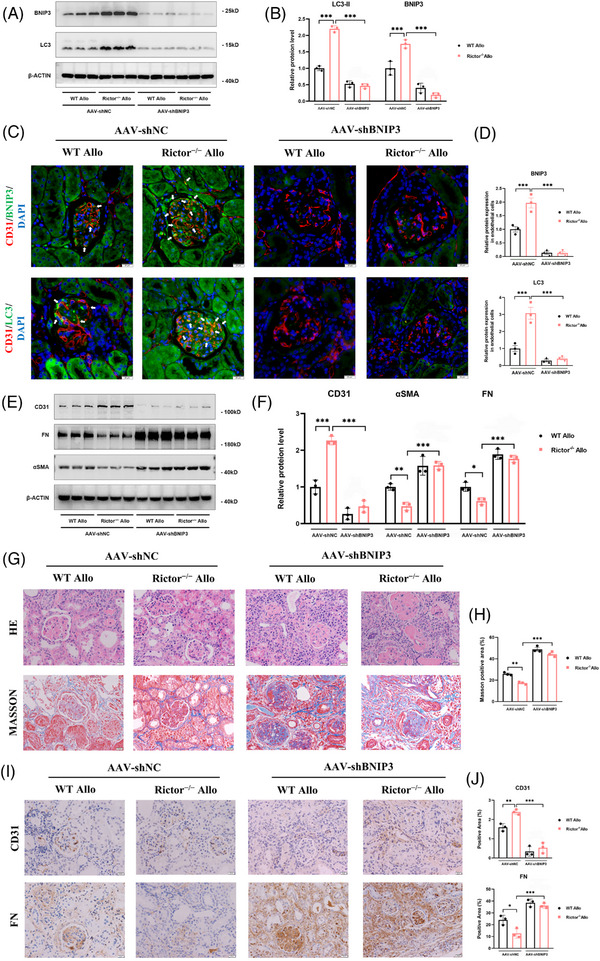
Our findings revealed a significant activation of the Rictor/mTORC2 signalling in CAD patients and allogeneic kidney transplanted mice. The suppression of Rictor/mTORC2 signalling alleviated TNFα-induced EndMT in HUVECs. Moreover, Rictor knockout in endothelial cells remarkably ameliorated renal vascular EndMT and allograft interstitial fibrosis in allogeneic kidney transplanted mice. Mechanistically, Rictor knockout resulted in an augmented BNIP3-mediated mitophagy in endothelial cells. Furthermore, Rictor/mTORC2 facilitated the MARCH5-mediated degradation of BNIP3 at the K130 site through K48-linked ubiquitination, thereby regulating mitophagy activity. Subsequent experiments also demonstrated that BNIP3 knockdown nearly reversed the enhanced mitophagy and mitigated EndMT and allograft interstitial fibrosis induced by Rictor knockout.
Consequently, our study underscores Rictor/mTORC2 signalling as a critical mediator of renal vascular EndMT and allograft interstitial fibrosis progression, exerting its impact through regulating BNIP3-mediated mitophagy. This insight unveils a potential therapeutic target for mitigating renal allograft interstitial fibrosis.
肾移植患者慢性移植物功能障碍(CAD)的主要组织病理学特征是肾移植间质纤维化/肾小管萎缩(IF/TA)。虽然肾血管内皮-间充质转化(EndMT)已被证实是 CAD 患者 IF/TA 的一个重要促成因素,但其潜在机制尚不清楚。通过单细胞转录组分析,我们发现 Rictor 可能是 EndMT 的一个潜在关键介质。本研究旨在阐明 Rictor/mTORC2 信号通路在肾移植间质纤维化发病机制中的作用及其相关机制。
通过细胞实验和肾同种异体移植小鼠模型中 Rictor 的耗竭,研究 Rictor/mTORC2 通路对肾血管 EndMT 和肾移植纤维化的影响。随后,进行了一系列实验来探讨 Rictor 敲除导致增强的线粒体自噬和改善的 EndMT 的潜在机制。
我们的研究结果表明,CAD 患者和同种异体肾移植小鼠中 Rictor/mTORC2 信号显著激活。抑制 Rictor/mTORC2 信号可减轻 TNFα诱导的 HUVECs 中的 EndMT。此外,内皮细胞中 Rictor 的敲除显著改善了同种异体肾移植小鼠的肾血管 EndMT 和移植间质纤维化。机制上,Rictor 敲除导致内皮细胞中 BNIP3 介导的线粒体自噬增加。此外,Rictor/mTORC2 通过 K48 连接的泛素化促进 MARCH5 介导的 BNIP3 在 K130 位点的降解,从而调节线粒体自噬活性。随后的实验还表明,BNIP3 敲低几乎逆转了 Rictor 敲除引起的增强的线粒体自噬,并减轻了 EndMT 和移植间质纤维化。
因此,我们的研究强调了 Rictor/mTORC2 信号作为肾血管 EndMT 和移植间质纤维化进展的关键介质,通过调节 BNIP3 介导的线粒体自噬发挥作用。这一发现为减轻肾移植间质纤维化提供了一个潜在的治疗靶点。