Structural Biology Initiative, CUNY Advanced Science Research Center, New York, New York, USA.
PhD Program in Biochemistry, CUNY Graduate Center, New York, New York, USA.
Protein Sci. 2024 Jun;33(6):e5024. doi: 10.1002/pro.5024.
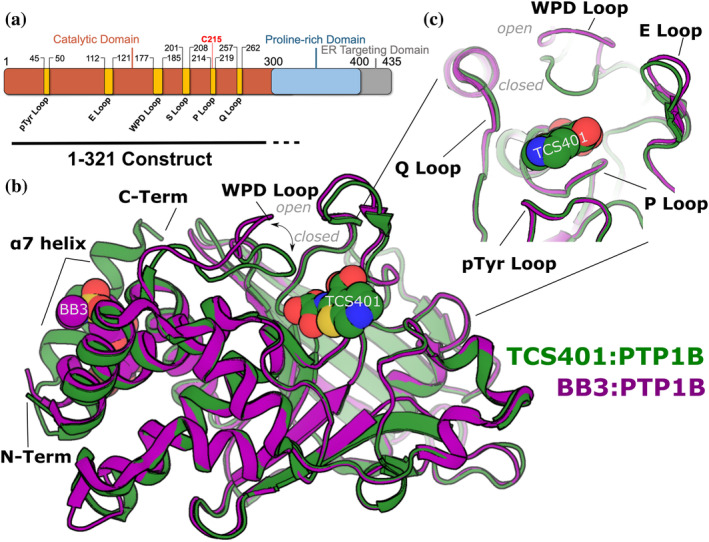
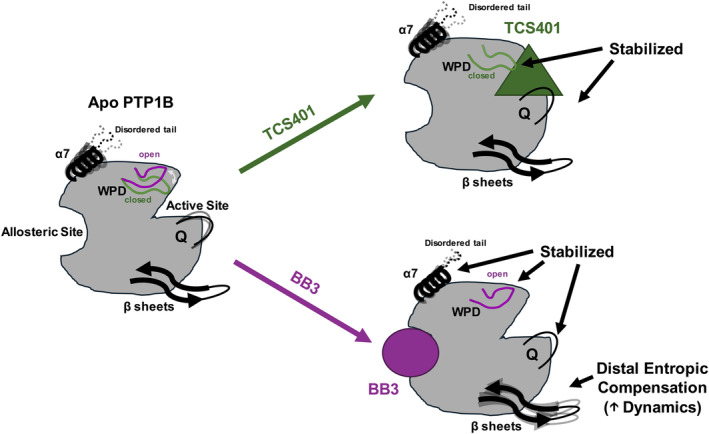
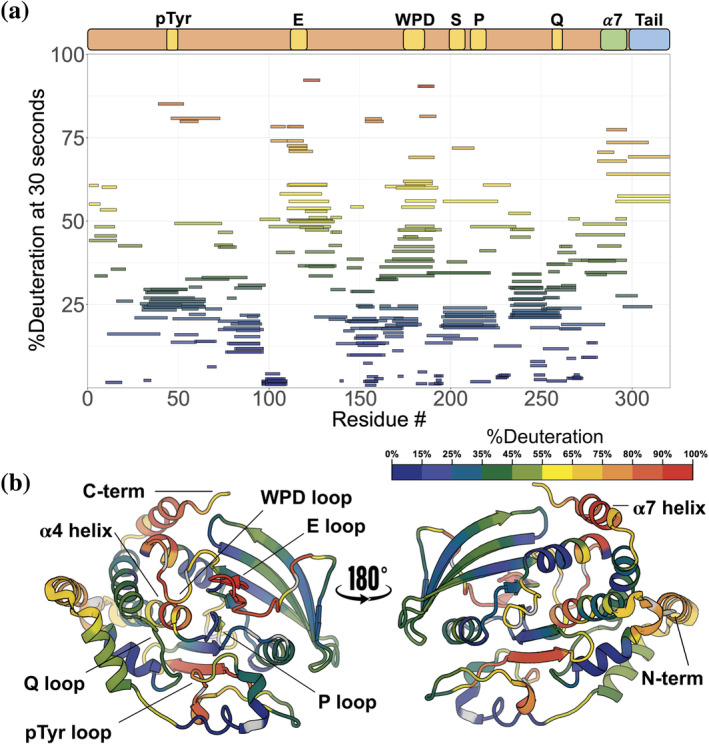
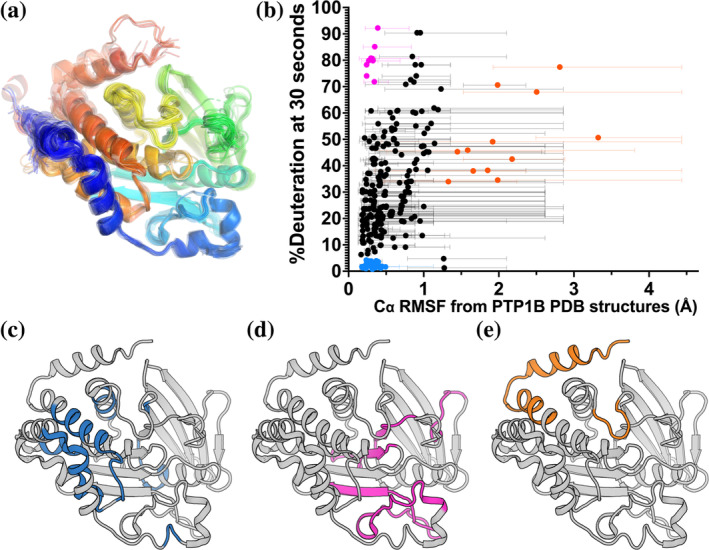
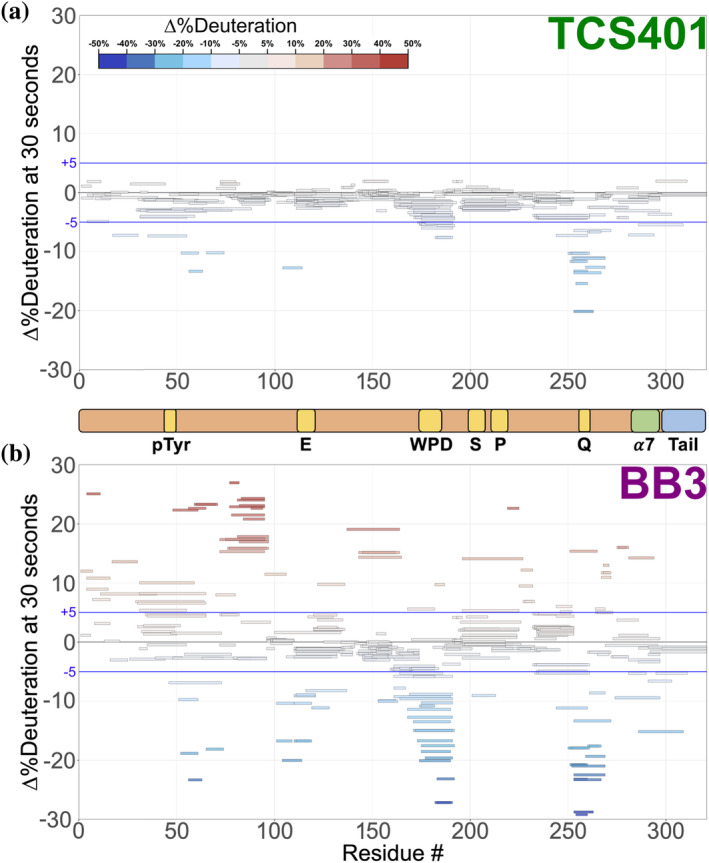
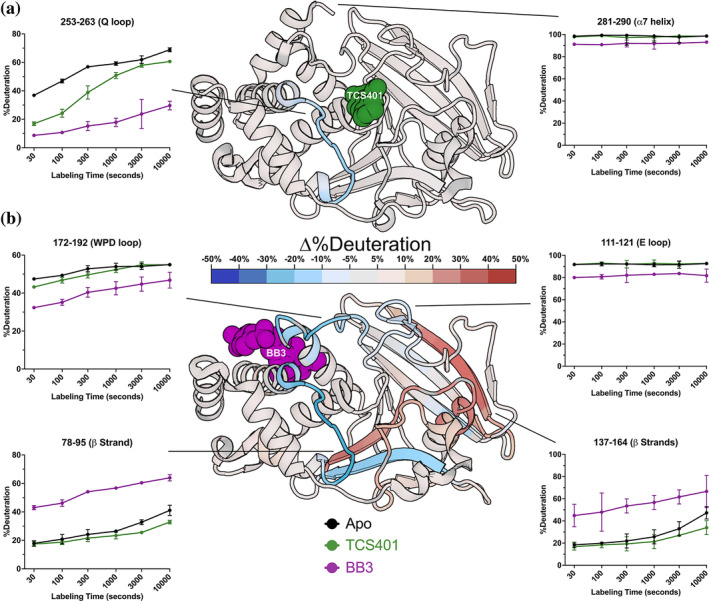
Protein tyrosine phosphatase 1B (PTP1B) is a validated therapeutic target for obesity, diabetes, and certain types of cancer. In particular, allosteric inhibitors hold potential for therapeutic use, but an incomplete understanding of conformational dynamics and allostery in this protein has hindered their development. Here, we interrogate solution dynamics and allosteric responses in PTP1B using high-resolution hydrogen-deuterium exchange mass spectrometry (HDX-MS), an emerging and powerful biophysical technique. Using HDX-MS, we obtain a detailed map of backbone amide exchange that serves as a proxy for the solution dynamics of apo PTP1B, revealing several flexible loops interspersed among more constrained and rigid regions within the protein structure, as well as local regions that exchange faster than expected from their secondary structure and solvent accessibility. We demonstrate that our HDX rate data obtained in solution adds value to estimates of conformational heterogeneity derived from a pseudo-ensemble constructed from ~200 crystal structures of PTP1B. Furthermore, we report HDX-MS maps for PTP1B with active-site versus allosteric small-molecule inhibitors. These maps suggest distinct and widespread effects on protein dynamics relative to the apo form, including changes in locations distal (>35 Å) from the respective ligand binding sites. These results illuminate that allosteric inhibitors of PTP1B can induce unexpected changes in dynamics that extend beyond the previously understood allosteric network. Together, our data suggest a model of BB3 allostery in PTP1B that combines conformational restriction of active-site residues with compensatory liberation of distal residues that aid in entropic balancing. Overall, our work showcases the potential of HDX-MS for elucidating aspects of protein conformational dynamics and allosteric effects of small-molecule ligands and highlights the potential of integrating HDX-MS alongside other complementary methods, such as room-temperature X-ray crystallography, NMR spectroscopy, and molecular dynamics simulations, to guide the development of new therapeutics.
蛋白酪氨酸磷酸酶 1B(PTP1B)是肥胖症、糖尿病和某些类型癌症的一种经过验证的治疗靶标。特别是,变构抑制剂具有治疗用途的潜力,但对该蛋白质构象动力学和变构作用的理解不完整,阻碍了它们的发展。在这里,我们使用高分辨率氢氘交换质谱(HDX-MS)来研究 PTP1B 中的溶液动力学和变构反应,这是一种新兴的强大生物物理技术。使用 HDX-MS,我们获得了一个详细的骨架酰胺交换图谱,该图谱可作为 apo PTP1B 溶液动力学的替代物,揭示了几个灵活的环散布在蛋白质结构中更受约束和刚性的区域之间,以及一些局部区域的交换速度快于其二级结构和溶剂可及性的预期。我们证明,我们在溶液中获得的 HDX 速率数据增加了源自 PTP1B 的~200 个晶体结构构建的伪系综中得出的构象异质性估计值的价值。此外,我们报告了 PTP1B 的活性位点与变构小分子抑制剂的 HDX-MS 图谱。这些图谱表明,与 apo 形式相比,对蛋白质动力学具有明显且广泛的影响,包括相对于各自的配体结合位点的位置变化(>35Å)。这些结果表明,PTP1B 的变构抑制剂可以诱导超出先前理解的变构网络的意想不到的动力学变化。总之,我们的数据表明,PTP1B 中 BB3 变构的模型结合了活性位点残基的构象限制与远端残基的补偿释放,这有助于熵平衡。总的来说,我们的工作展示了 HDX-MS 阐明蛋白质构象动力学和小分子配体变构效应的潜力,并强调了将 HDX-MS 与其他互补方法(如室温 X 射线晶体学、NMR 光谱学和分子动力学模拟)相结合的潜力,以指导新疗法的开发。