Department of Epidemiology and Biostatistics, University of Arizona Mel and Enid Zuckerman College of Public Health, Tucson, AZ, USA.
University of Arizona Cancer Center, University of Arizona, Tucson, AZ, USA.
NPJ Syst Biol Appl. 2024 May 28;10(1):58. doi: 10.1038/s41540-024-00386-w.
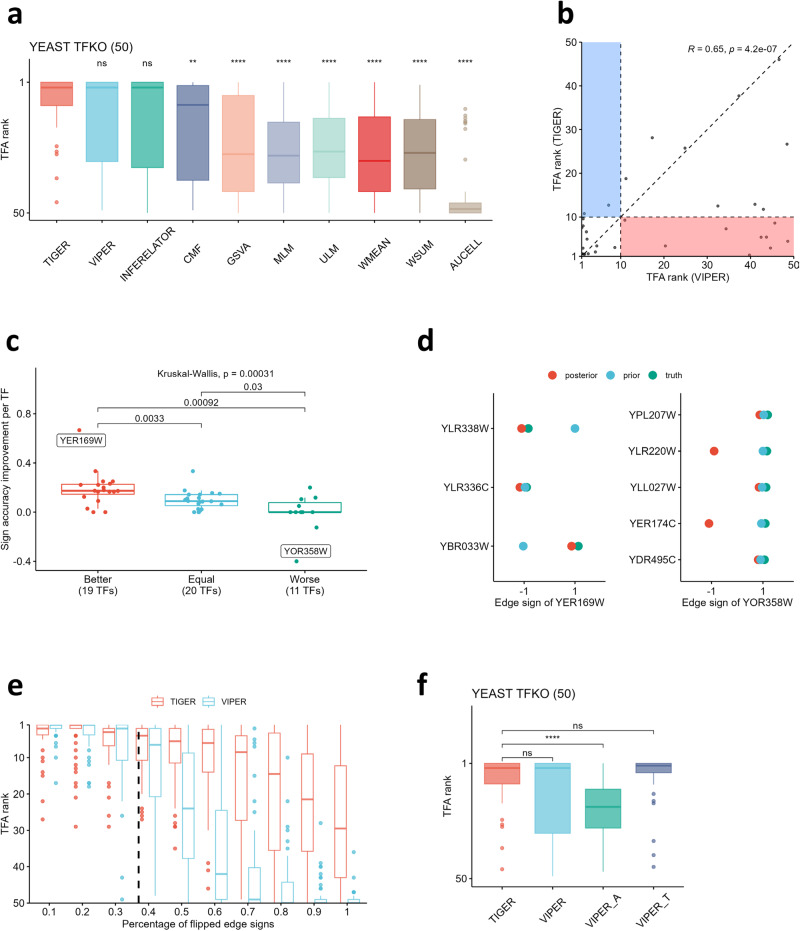
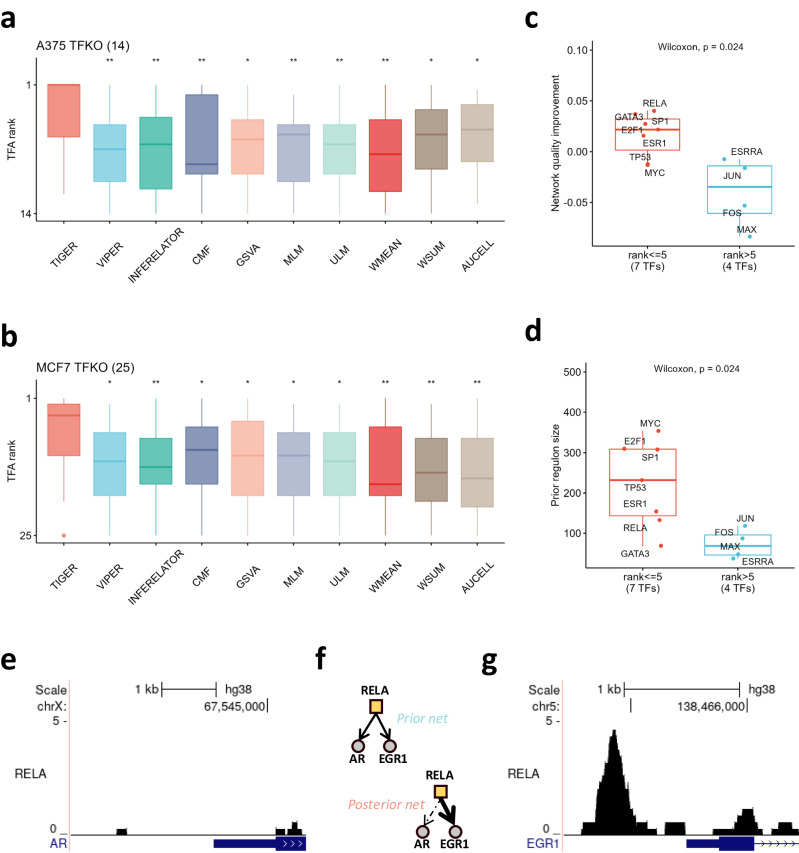
Transcriptional regulation plays a crucial role in determining cell fate and disease, yet inferring the key regulators from gene expression data remains a significant challenge. Existing methods for estimating transcription factor (TF) activity often rely on static TF-gene interaction databases and cannot adapt to changes in regulatory mechanisms across different cell types and disease conditions. Here, we present a new algorithm - Transcriptional Inference using Gene Expression and Regulatory data (TIGER) - that overcomes these limitations by flexibly modeling activation and inhibition events, up-weighting essential edges, shrinking irrelevant edges towards zero through a sparse Bayesian prior, and simultaneously estimating both TF activity levels and changes in the underlying regulatory network. When applied to yeast and cancer TF knock-out datasets, TIGER outperforms comparable methods in terms of prediction accuracy. Moreover, our application of TIGER to tissue- and cell-type-specific RNA-seq data demonstrates its ability to uncover differences in regulatory mechanisms. Collectively, our findings highlight the utility of modeling context-specific regulation when inferring transcription factor activities.
转录调控在决定细胞命运和疾病方面起着至关重要的作用,但从基因表达数据中推断关键调控因子仍然是一个重大挑战。现有的估计转录因子 (TF) 活性的方法通常依赖于静态的 TF-基因相互作用数据库,并且不能适应不同细胞类型和疾病条件下调控机制的变化。在这里,我们提出了一种新的算法——使用基因表达和调控数据进行转录推断 (TIGER)——通过灵活地建模激活和抑制事件、对重要边进行加权、通过稀疏贝叶斯先验将不相关的边收缩到零,同时估计 TF 活性水平和基础调控网络的变化,克服了这些限制。当应用于酵母和癌症 TF 敲除数据集时,TIGER 在预测准确性方面优于可比方法。此外,我们将 TIGER 应用于组织和细胞类型特异性 RNA-seq 数据,证明了它能够揭示调控机制的差异。总之,我们的研究结果强调了在推断转录因子活性时,对上下文特定调控进行建模的实用性。