Badia-I-Mompel Pau, Vélez Santiago Jesús, Braunger Jana, Geiss Celina, Dimitrov Daniel, Müller-Dott Sophia, Taus Petr, Dugourd Aurelien, Holland Christian H, Ramirez Flores Ricardo O, Saez-Rodriguez Julio
Heidelberg University, Faculty of Medicine, and Heidelberg University Hospital, Institute for Computational Biomedicine, BioQuant, Heidelberg 69120, Germany.
Institute for Computational Biomedicine, Heidelberg University Hospital, BioQuant, Heidelberg 69120, Germany.
Bioinform Adv. 2022 Mar 8;2(1):vbac016. doi: 10.1093/bioadv/vbac016. eCollection 2022.
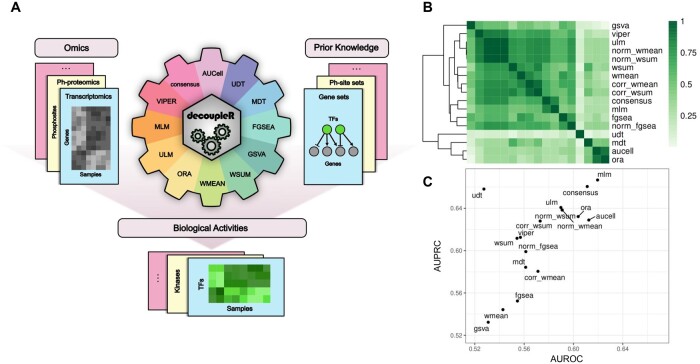
Many methods allow us to extract biological activities from omics data using information from prior knowledge resources, reducing the dimensionality for increased statistical power and better interpretability. Here, we present decoupleR, a Bioconductor and Python package containing computational methods to extract these activities within a unified framework. decoupleR allows us to flexibly run any method with a given resource, including methods that leverage mode of regulation and weights of interactions, which are not present in other frameworks. Moreover, it leverages OmniPath, a meta-resource comprising over 100 databases of prior knowledge. Using decoupleR, we evaluated the performance of methods on transcriptomic and phospho-proteomic perturbation experiments. Our findings suggest that simple linear models and the consensus score across top methods perform better than other methods at predicting perturbed regulators.
decoupleR's open-source code is available in Bioconductor (https://www.bioconductor.org/packages/release/bioc/html/decoupleR.html) for R and in GitHub (https://github.com/saezlab/decoupler-py) for Python. The code to reproduce the results is in GitHub (https://github.com/saezlab/decoupleR_manuscript) and the data in Zenodo (https://zenodo.org/record/5645208).
Supplementary data are available at online.
许多方法可让我们利用先验知识资源中的信息从组学数据中提取生物学活性,降低维度以提高统计功效和增强可解释性。在此,我们展示了decoupleR,这是一个包含计算方法的Bioconductor和Python软件包,用于在统一框架内提取这些活性。decoupleR使我们能够灵活地使用给定资源运行任何方法,包括利用调控模式和相互作用权重的方法,而其他框架中不存在这些方法。此外,它利用了OmniPath,这是一个包含100多个先验知识数据库的元资源。使用decoupleR,我们评估了这些方法在转录组学和磷酸化蛋白质组学扰动实验中的性能。我们的研究结果表明,简单线性模型和顶级方法的共识评分在预测受扰动的调节因子方面比其他方法表现更好。
decoupleR的开源代码可在Bioconductor(https://www.bioconductor.org/packages/release/bioc/html/decoupleR.html)上获取用于R,在GitHub(https://github.com/saezlab/decoupler-py)上获取用于Python。重现结果的代码在GitHub(https://github.com/saezlab/decoupleR_manuscript)中,数据在Zenodo(https://zenodo.org/record/5645208)中。
补充数据可在网上获取。