Department of Chemical Engineering, Stanford University School, Stanford, CA, USA.
Stanford Data Science, Stanford University, Stanford, CA, USA.
Nat Protoc. 2024 Aug;19(8):2283-2297. doi: 10.1038/s41596-024-00991-3. Epub 2024 Jun 6.
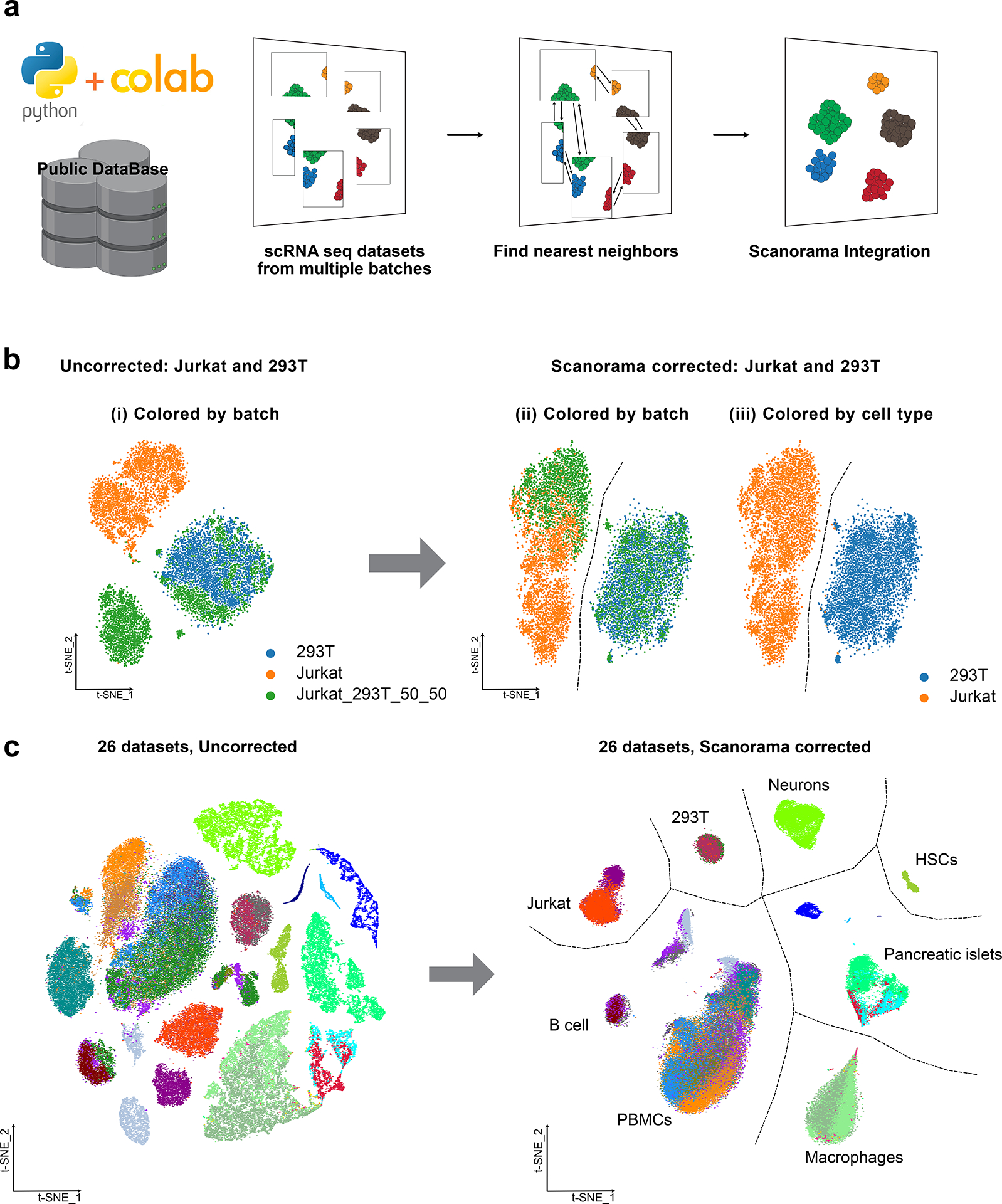
Merging diverse single-cell RNA sequencing (scRNA-seq) data from numerous experiments, laboratories and technologies can uncover important biological insights. Nonetheless, integrating scRNA-seq data encounters special challenges when the datasets are composed of diverse cell type compositions. Scanorama offers a robust solution for improving the quality and interpretation of heterogeneous scRNA-seq data by effectively merging information from diverse sources. Scanorama is designed to address the technical variation introduced by differences in sample preparation, sequencing depth and experimental batches that can confound the analysis of multiple scRNA-seq datasets. Here we provide a detailed protocol for using Scanorama within a Scanpy-based single-cell analysis workflow coupled with Google Colaboratory, a cloud-based free Jupyter notebook environment service. The protocol involves Scanorama integration, a process that typically spans 0.5-3 h. Scanorama integration requires a basic understanding of cellular biology, transcriptomic technologies and bioinformatics. Our protocol and new Scanorama-Colaboratory resource should make scRNA-seq integration more widely accessible to researchers.
合并来自多个实验、实验室和技术的多样化单细胞 RNA 测序(scRNA-seq)数据,可以揭示重要的生物学见解。然而,当数据集由不同的细胞类型组成时,整合 scRNA-seq 数据会遇到特殊的挑战。Scanorama 通过有效合并来自不同来源的信息,为提高异质 scRNA-seq 数据的质量和解释提供了强大的解决方案。Scanorama 旨在解决由样本制备、测序深度和实验批次差异引入的技术差异,这些差异会混淆多个 scRNA-seq 数据集的分析。在这里,我们提供了一个在基于 Scanpy 的单细胞分析工作流程中使用 Scanorama 的详细协议,该工作流程与 Google Colaboratory 相结合,这是一个基于云的免费 Jupyter 笔记本环境服务。该协议涉及 Scanorama 集成,这一过程通常需要 0.5-3 小时。Scanorama 集成需要对细胞生物学、转录组学技术和生物信息学有基本的了解。我们的协议和新的 Scanorama-Colaboratory 资源应该使 scRNA-seq 集成更广泛地为研究人员所接受。