Hangzhou Institute of Medicine, Chinese Academy of Sciences, Hangzhou, 310018 Zhejiang, China.
College of Electrical and Information Engineering, Hunan University, Changsha, 410082 Hunan, China.
Brief Bioinform. 2024 May 23;25(4). doi: 10.1093/bib/bbae321.
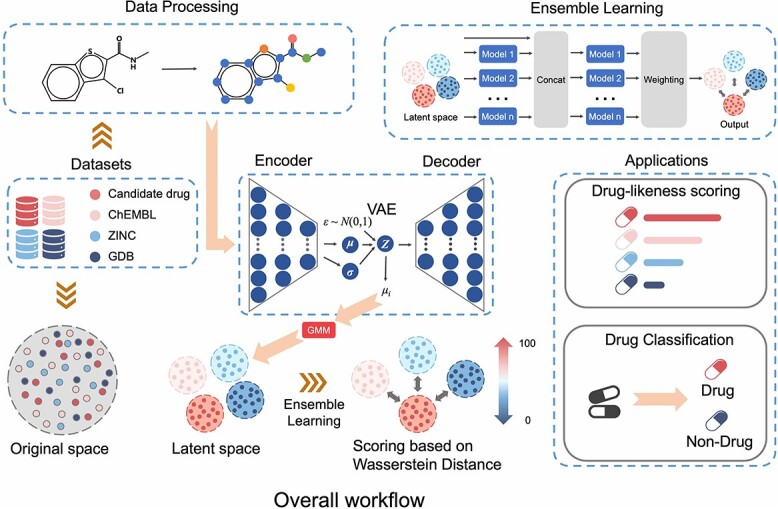
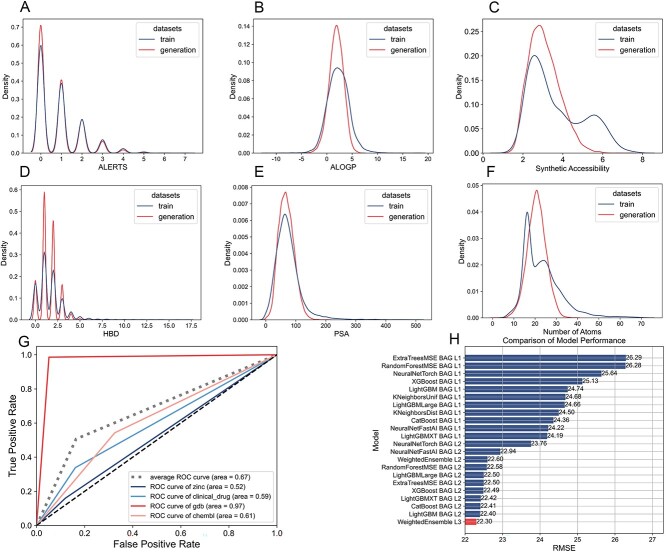
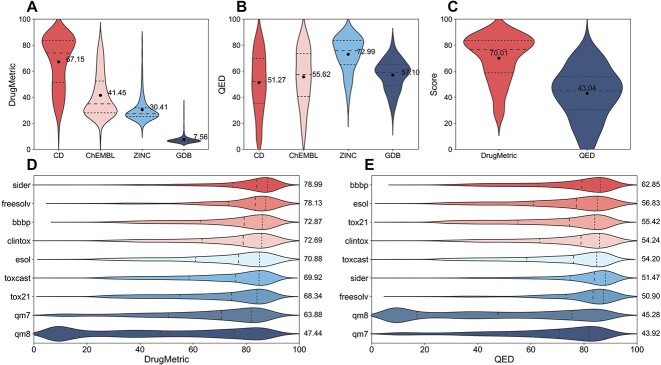
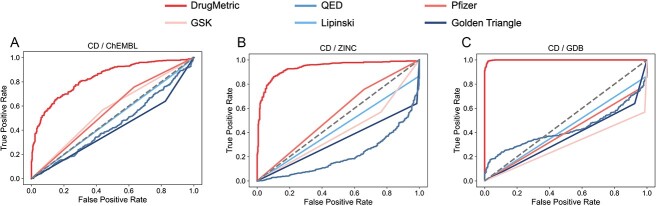
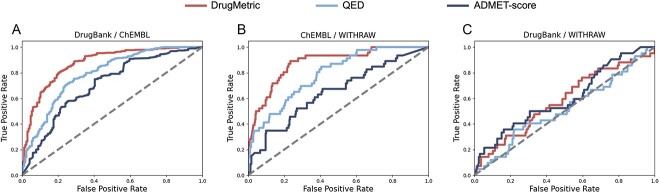
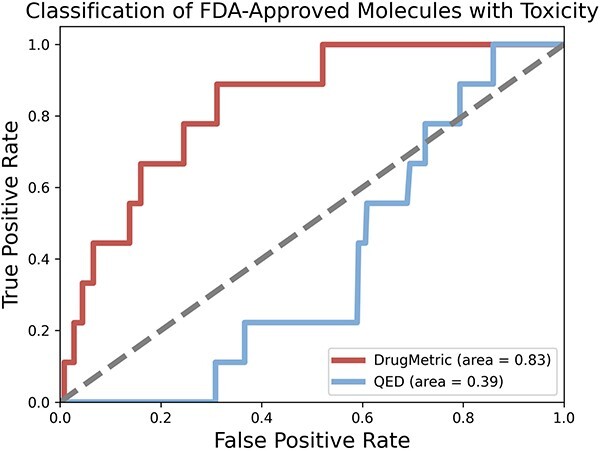
The process of drug discovery is widely known to be lengthy and resource-intensive. Artificial Intelligence approaches bring hope for accelerating the identification of molecules with the necessary properties for drug development. Drug-likeness assessment is crucial for the virtual screening of candidate drugs. However, traditional methods like Quantitative Estimation of Drug-likeness (QED) struggle to distinguish between drug and non-drug molecules accurately. Additionally, some deep learning-based binary classification models heavily rely on selecting training negative sets. To address these challenges, we introduce a novel unsupervised learning framework called DrugMetric, an innovative framework for quantitatively assessing drug-likeness based on the chemical space distance. DrugMetric blends the powerful learning ability of variational autoencoders with the discriminative ability of the Gaussian Mixture Model. This synergy enables DrugMetric to identify significant differences in drug-likeness across different datasets effectively. Moreover, DrugMetric incorporates principles of ensemble learning to enhance its predictive capabilities. Upon testing over a variety of tasks and datasets, DrugMetric consistently showcases superior scoring and classification performance. It excels in quantifying drug-likeness and accurately distinguishing candidate drugs from non-drugs, surpassing traditional methods including QED. This work highlights DrugMetric as a practical tool for drug-likeness scoring, facilitating the acceleration of virtual drug screening, and has potential applications in other biochemical fields.
药物发现的过程众所周知是漫长且资源密集的。人工智能方法为加速识别具有药物开发所需特性的分子带来了希望。药物相似性评估对于候选药物的虚拟筛选至关重要。然而,像定量药物相似性估计(QED)这样的传统方法很难准确区分药物和非药物分子。此外,一些基于深度学习的二进制分类模型严重依赖于选择训练负集。为了解决这些挑战,我们引入了一种名为 DrugMetric 的新型无监督学习框架,这是一种基于化学空间距离的定量药物相似性评估的创新框架。DrugMetric 将变分自动编码器的强大学习能力与高斯混合模型的判别能力相结合。这种协同作用使 DrugMetric 能够有效地识别不同数据集之间的药物相似性的显著差异。此外,DrugMetric 结合了集成学习的原则,以增强其预测能力。在对各种任务和数据集进行测试后,DrugMetric 始终展示出出色的评分和分类性能。它在量化药物相似性和准确区分候选药物与非药物方面表现出色,优于包括 QED 在内的传统方法。这项工作突出了 DrugMetric 作为药物相似性评分的实用工具,加速了虚拟药物筛选,并在其他生化领域具有潜在的应用。