Wang Yi, Woyshner Kyla, Sriworarat Chaichontat, Stein-O'Brien Genevieve, Goff Loyal A, Hansen Kasper D
Department of Biostatistics, Johns Hopkins Bloomberg School of Public Health.
Department of Genetic Medicine, Johns Hopkins School of Medicine.
bioRxiv. 2024 Nov 27:2024.07.01.599554. doi: 10.1101/2024.07.01.599554.
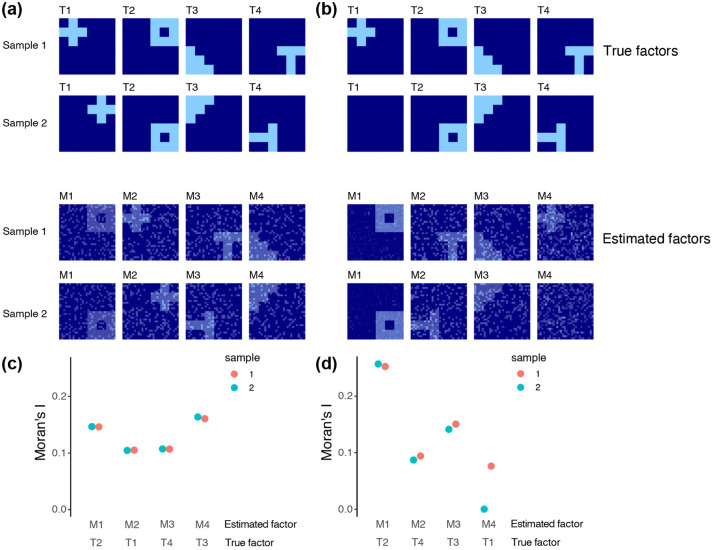
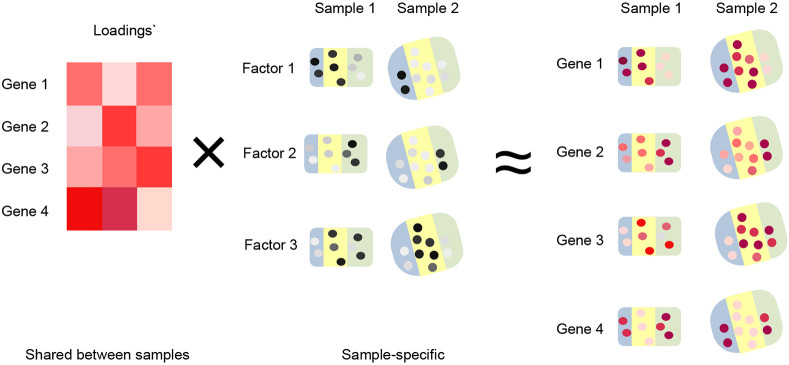
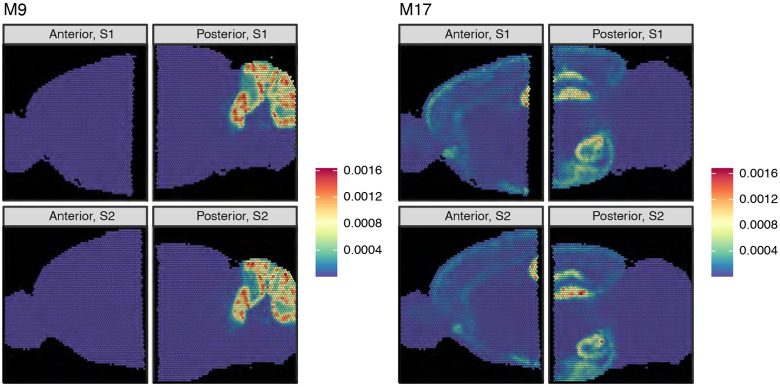
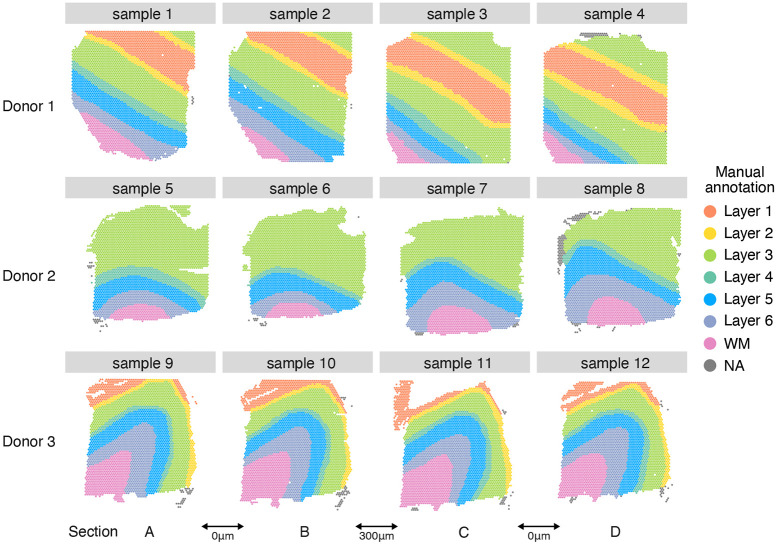
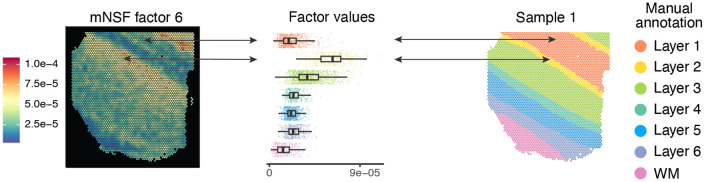
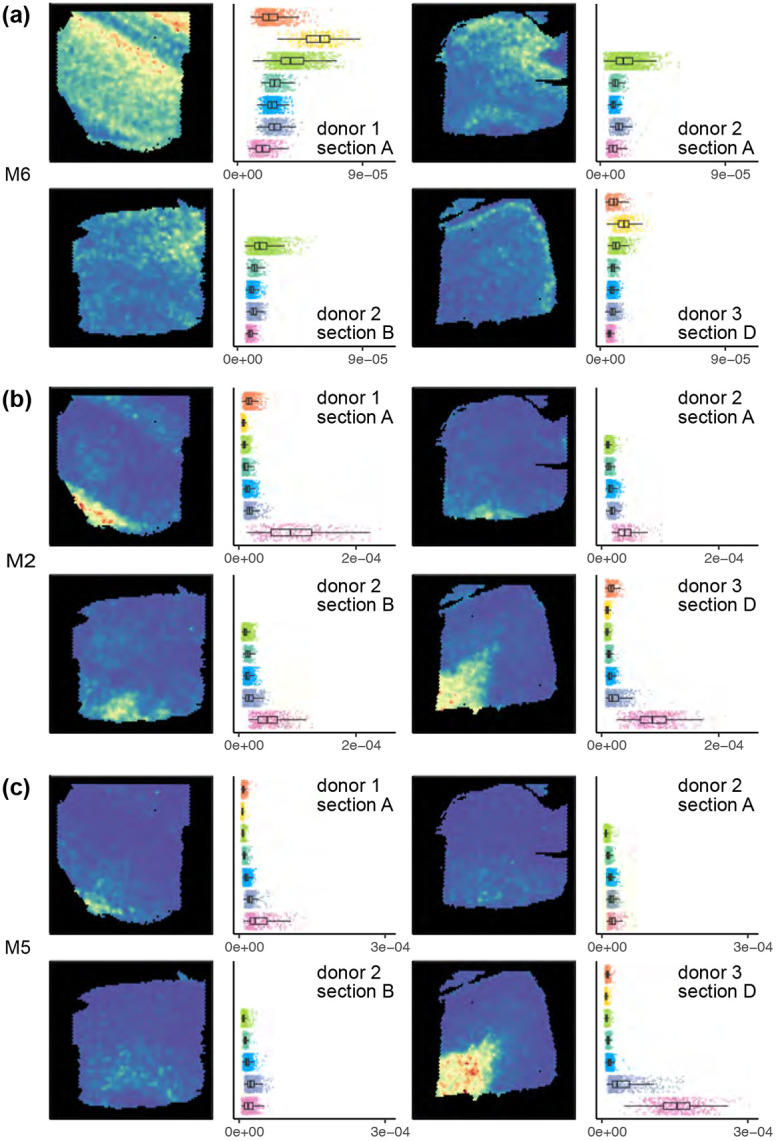
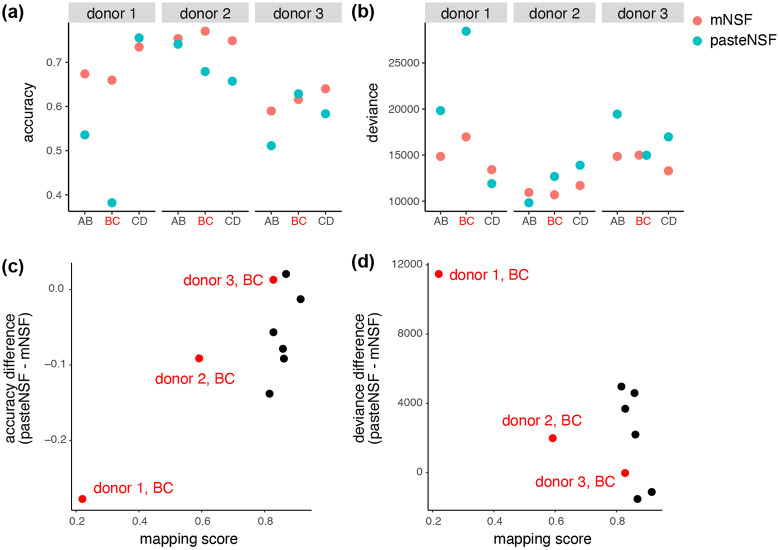
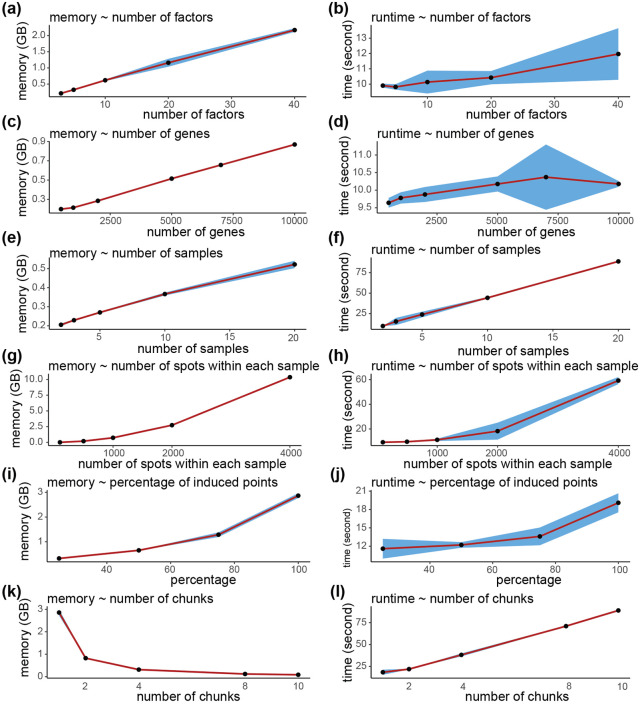
Analyzing multi-sample spatial transcriptomics data requires accounting for biological variation. We present multi-sample non-negative spatial factorization (mNSF), an alignment-free framework extending single-sample spatial factorization (NSF) to multi-sample datasets. mNSF incorporates sample-specific spatial correlation modeling and extracts low-dimensional data representations. Through simulations and real data analysis, we demonstrate mNSF's efficacy in identifying true factors, shared anatomical regions, and region-specific biological functions. mNSF's performance is comparable to alignment-based methods when alignment is feasible, while enabling analysis in scenarios where spatial alignment is unfeasible. mNSF shows promise as a robust method for analyzing spatially resolved transcriptomics data across multiple samples.
分析多样本空间转录组学数据需要考虑生物学变异。我们提出了多样本非负空间因子分解(mNSF),这是一个无比对框架,将单样本空间因子分解(NSF)扩展到多样本数据集。mNSF纳入了样本特异性空间相关性建模,并提取低维数据表示。通过模拟和实际数据分析,我们证明了mNSF在识别真实因子、共享解剖区域和区域特异性生物学功能方面的有效性。在可行比对的情况下,mNSF的性能与基于比对的方法相当,同时能够在空间比对不可行的情况下进行分析。mNSF有望成为一种强大的方法,用于分析多个样本的空间分辨转录组学数据。