Department of Biology, Pennsylvania State University, University Park, PA 16802, USA.
Department of Biology and Ecology, University of Ostrava, Ostrava 701 03, Czechia.
Genetics. 2024 Sep 4;228(1). doi: 10.1093/genetics/iyae110.
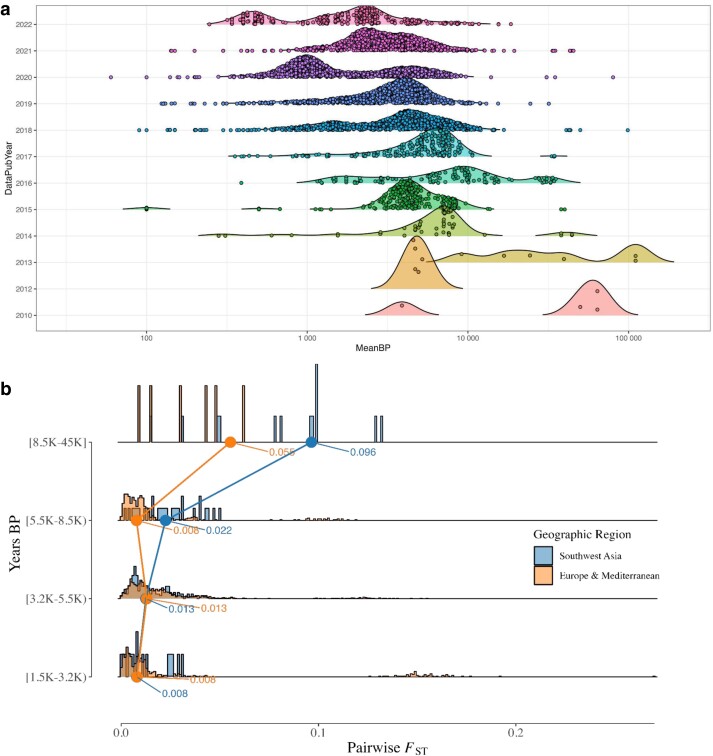
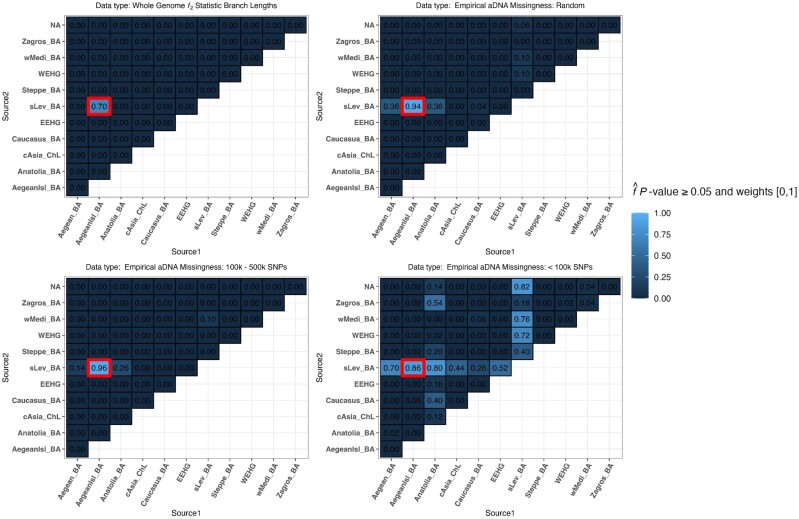
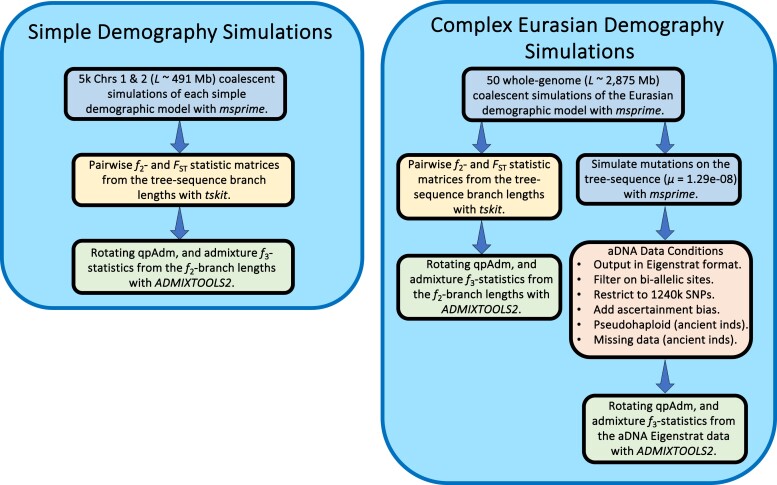
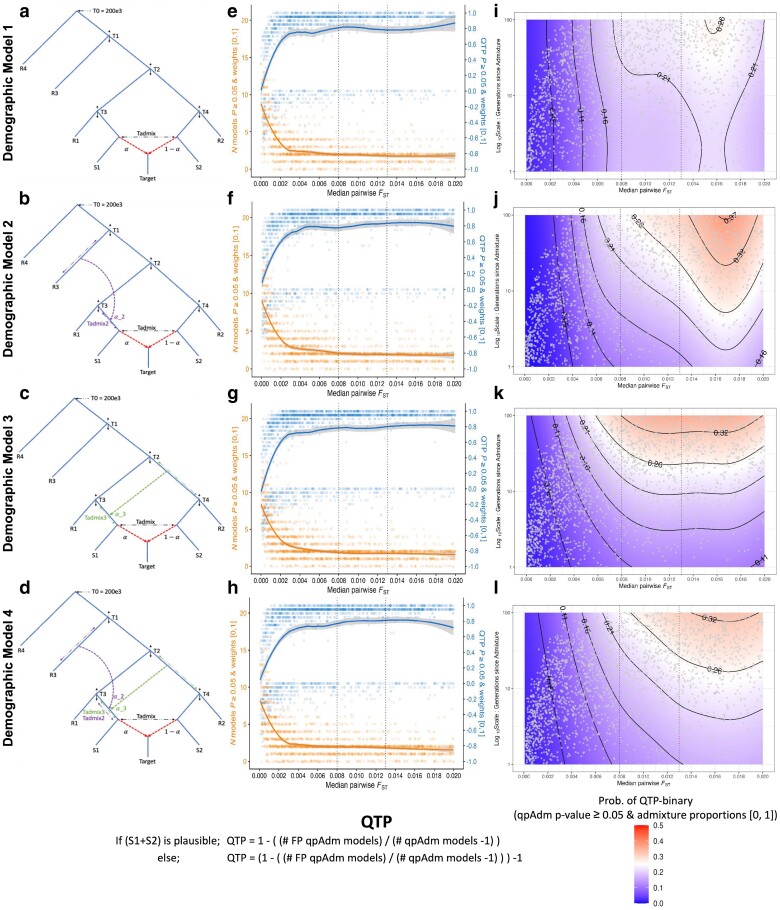
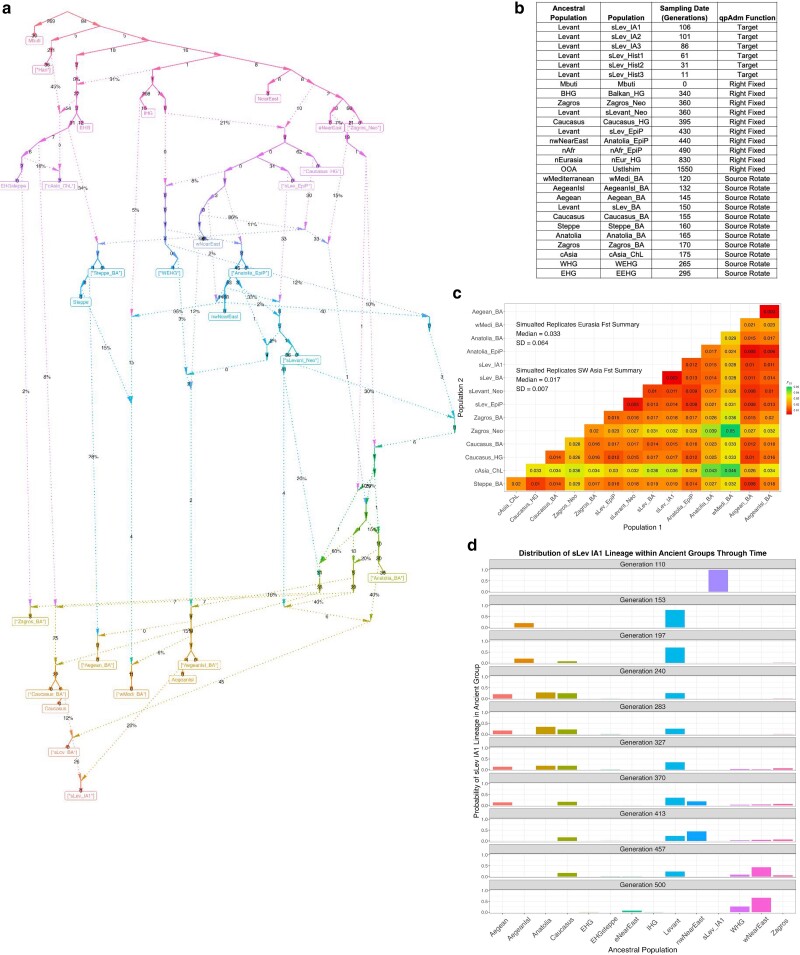
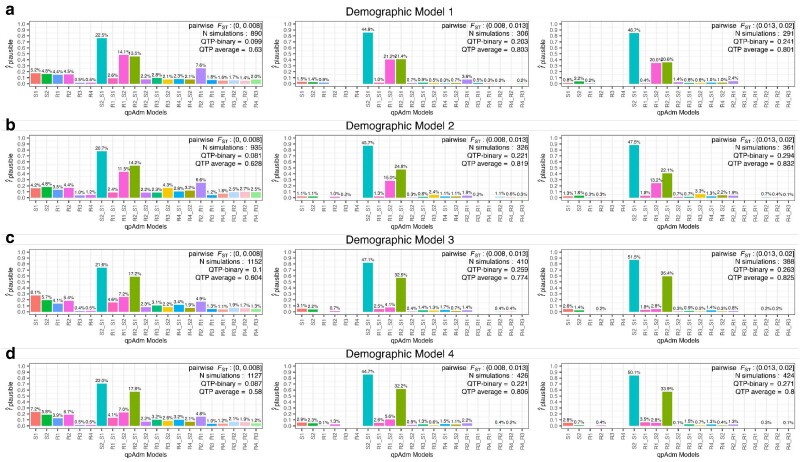
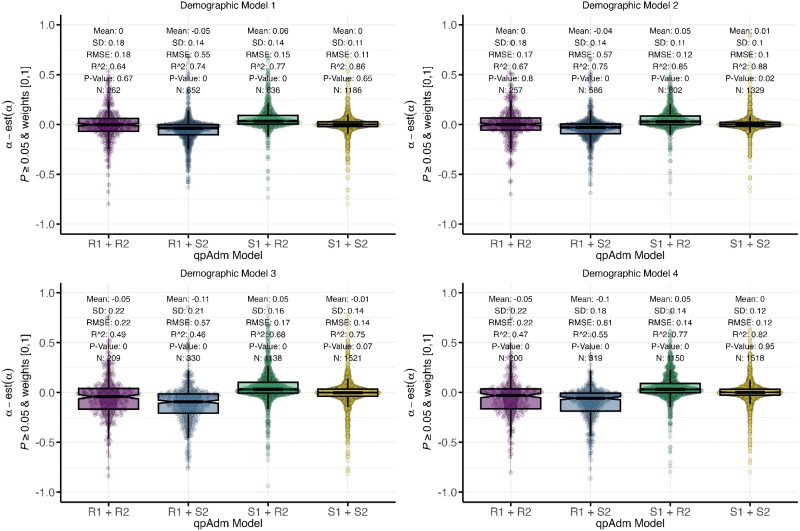
Our knowledge of human evolutionary history has been greatly advanced by paleogenomics. Since the 2020s, the study of ancient DNA has increasingly focused on reconstructing the recent past. However, the accuracy of paleogenomic methods in resolving questions of historical and archaeological importance amidst the increased demographic complexity and decreased genetic differentiation remains an open question. We evaluated the performance and behavior of two commonly used methods, qpAdm and the f3-statistic, on admixture inference under a diversity of demographic models and data conditions. We performed two complementary simulation approaches-firstly exploring a wide demographic parameter space under four simple demographic models of varying complexities and configurations using branch-length data from two chromosomes-and secondly, we analyzed a model of Eurasian history composed of 59 populations using whole-genome data modified with ancient DNA conditions such as SNP ascertainment, data missingness, and pseudohaploidization. We observe that population differentiation is the primary factor driving qpAdm performance. Notably, while complex gene flow histories influence which models are classified as plausible, they do not reduce overall performance. Under conditions reflective of the historical period, qpAdm most frequently identifies the true model as plausible among a small candidate set of closely related populations. To increase the utility for resolving fine-scaled hypotheses, we provide a heuristic for further distinguishing between candidate models that incorporates qpAdm model P-values and f3-statistics. Finally, we demonstrate a significant performance increase for qpAdm using whole-genome branch-length f2-statistics, highlighting the potential for improved demographic inference that could be achieved with future advancements in f-statistic estimations.
古基因组学极大地推动了我们对人类进化历史的认识。自 21 世纪 20 年代以来,古 DNA 的研究越来越关注重建近代历史。然而,古基因组学方法在解决历史和考古学问题时的准确性,在人口结构日益复杂和遗传分化程度降低的情况下,仍然是一个悬而未决的问题。我们评估了两种常用方法(qpAdm 和 f3 统计量)在不同人口统计学模型和数据条件下进行混合推断的性能和行为。我们采用了两种互补的模拟方法:首先,在四个不同复杂程度和配置的简单人口统计学模型下,利用来自两条染色体的分支长度数据,探索广泛的人口统计学参数空间;其次,我们使用全基因组数据分析了一个由 59 个群体组成的欧亚历史模型,该模型经过了 SNP 确定、数据缺失和伪单体化等古 DNA 条件的修改。我们观察到,群体分化是影响 qpAdm 性能的主要因素。值得注意的是,尽管复杂的基因流动历史影响了哪些模型被归类为合理的,但它们并没有降低整体性能。在反映历史时期的条件下,qpAdm 最常将真实模型识别为来自一小部分密切相关群体的合理候选模型之一。为了提高解决精细尺度假说的能力,我们提供了一种启发式方法,用于进一步区分候选模型,该方法结合了 qpAdm 模型 P 值和 f3 统计量。最后,我们展示了使用全基因组分支长度 f2 统计量的 qpAdm 性能的显著提高,这突显了未来在 f 统计量估计方面取得进展,可能实现更好的人口统计学推断的潜力。