Behara Pavan Kumar, Jang Hyesu, Horton Joshua T, Gokey Trevor, Dotson David L, Boothroyd Simon, Bayly Christopher I, Cole Daniel J, Wang Lee-Ping, Mobley David L
Center for Neurotherapeutics, University of California, Irvine, California 92697, United States.
Chemistry Department, University of California at Davis, Davis, California 95616, United States.
J Phys Chem B. 2024 Aug 15;128(32):7888-7902. doi: 10.1021/acs.jpcb.4c03167. Epub 2024 Aug 1.
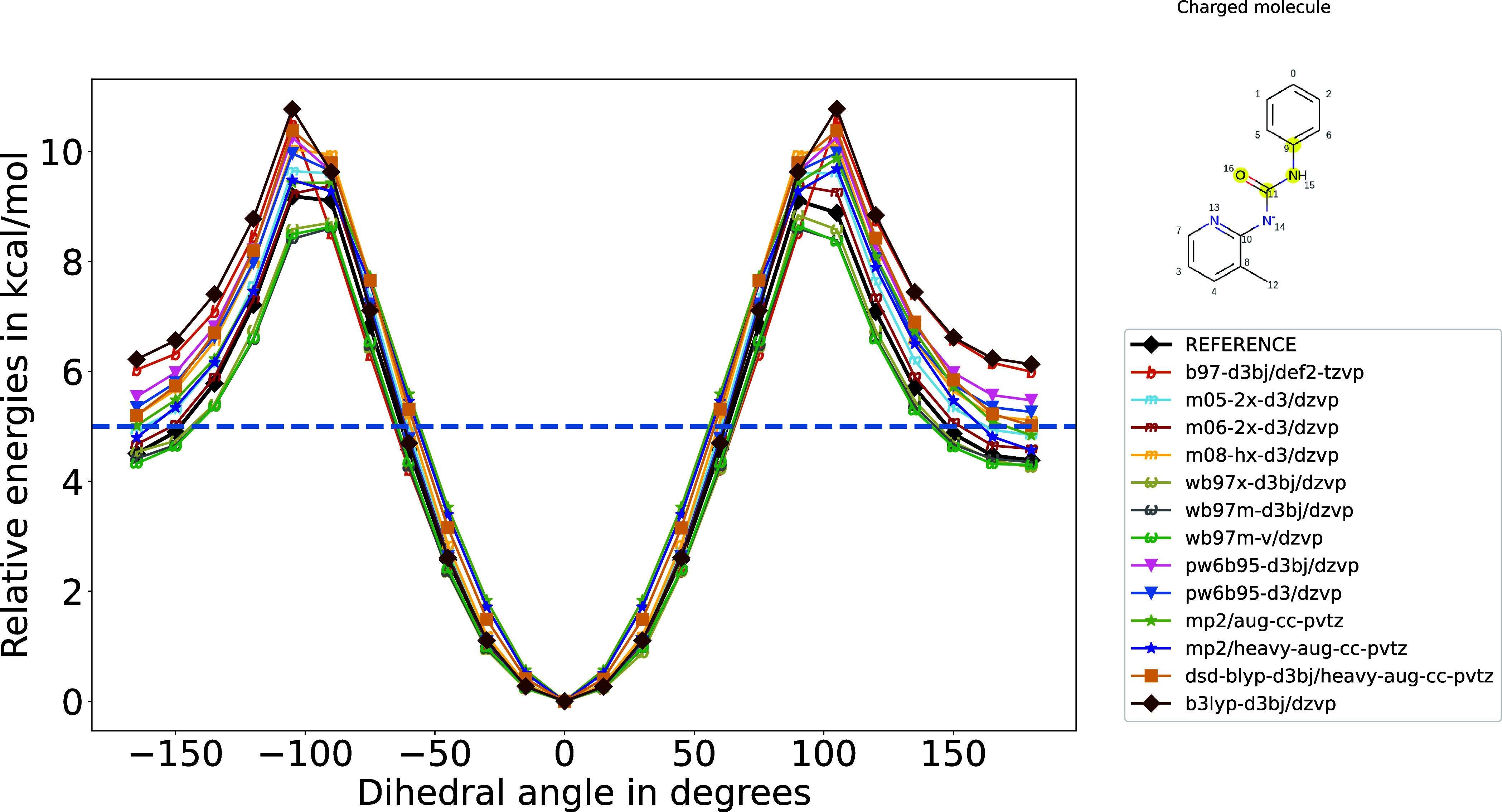
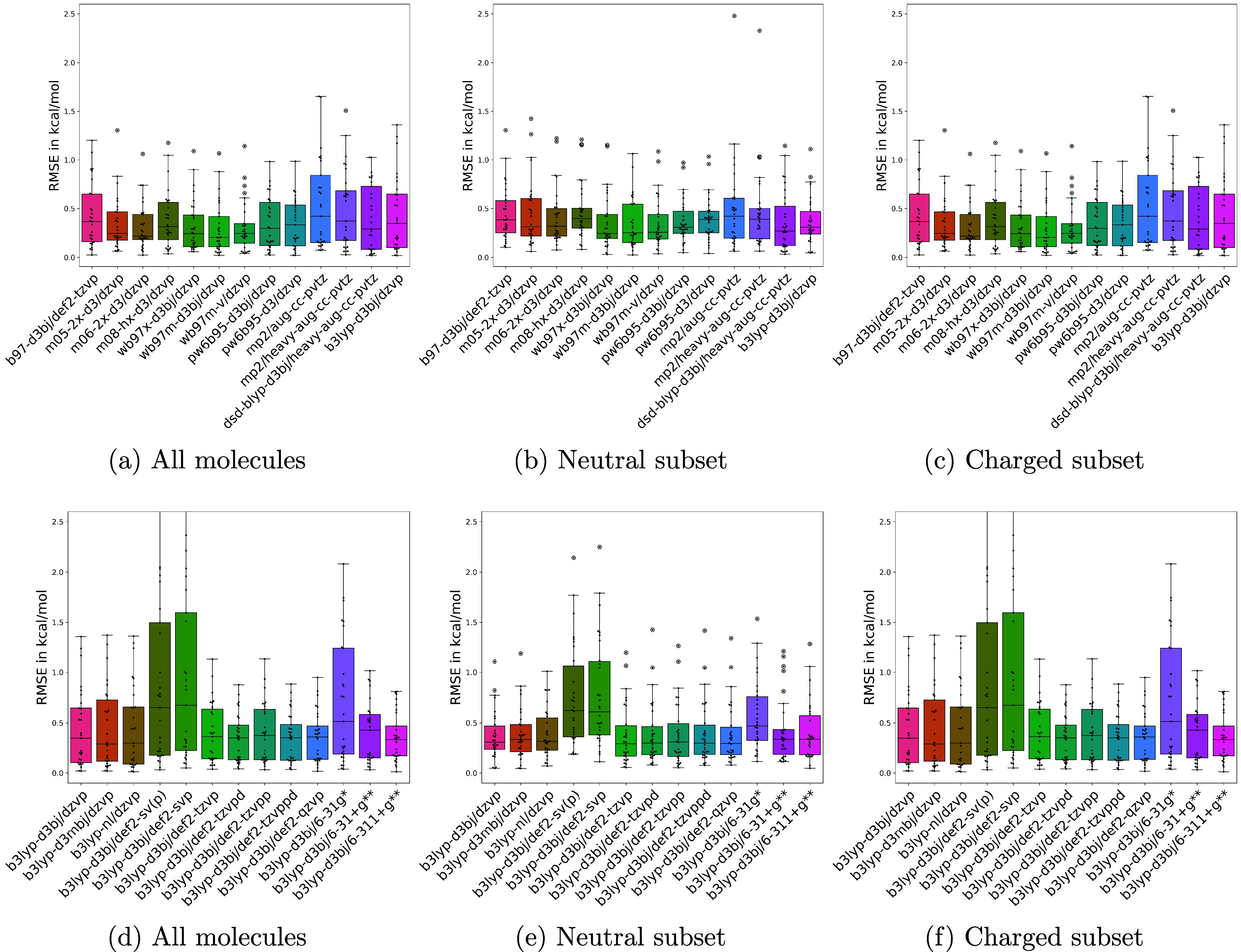
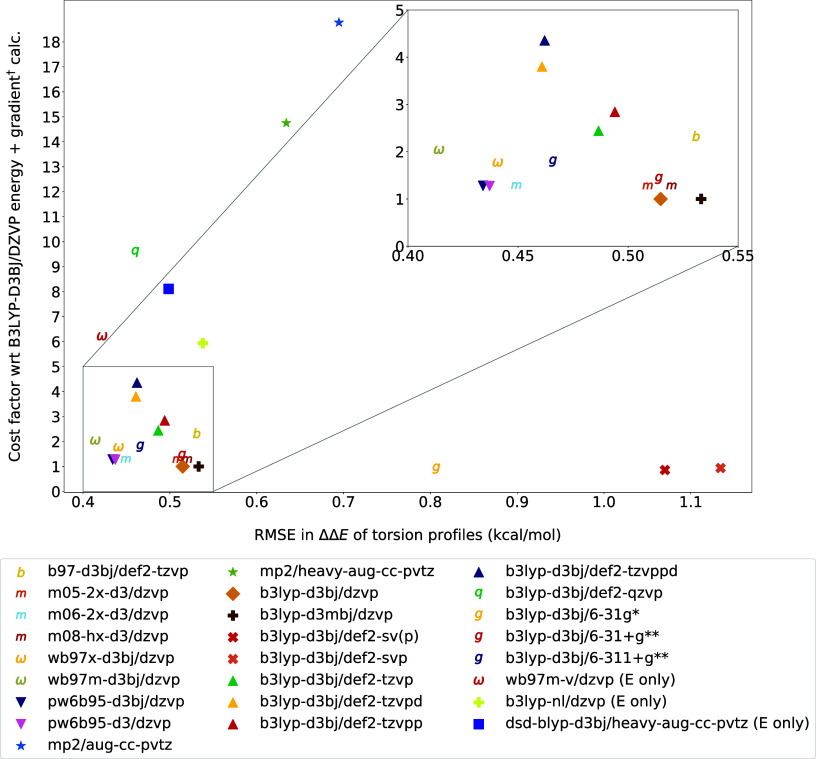
A wide range of density functional methods and basis sets are available to derive the electronic structure and properties of molecules. Quantum mechanical calculations are too computationally intensive for routine simulation of molecules in the condensed phase, prompting the development of computationally efficient force fields based on quantum mechanical data. Parametrizing general force fields, which cover a vast chemical space, necessitates the generation of sizable quantum mechanical data sets with optimized geometries and torsion scans. To achieve this efficiently, choosing a quantum mechanical method that balances computational cost and accuracy is crucial. In this study, we seek to assess the accuracy of quantum mechanical theory for specific properties such as conformer energies and torsion energetics. To comprehensively evaluate various methods, we focus on a representative set of 59 diverse small molecules, comparing approximately 25 combinations of functional and basis sets against the reference level coupled cluster calculations at the complete basis set limit.
有多种密度泛函方法和基组可用于推导分子的电子结构和性质。对于凝聚相中分子的常规模拟,量子力学计算的计算量太大,这促使人们基于量子力学数据开发计算效率高的力场。对涵盖广阔化学空间的通用力场进行参数化,需要生成具有优化几何结构和扭转扫描的大量量子力学数据集。为了高效地实现这一点,选择一种平衡计算成本和准确性的量子力学方法至关重要。在本研究中,我们试图评估量子力学理论对于诸如构象异构体能量和扭转能等特定性质的准确性。为了全面评估各种方法,我们聚焦于一组具有代表性的59个不同小分子,将大约25种泛函和基组的组合与完全基组极限下的参考水平耦合簇计算进行比较。