Crouzet Simon J, Lieberherr Anja Maria, Atz Kenneth, Nilsson Tobias, Sach-Peltason Lisa, Müller Alex T, Dal Peraro Matteo, Zhang Jitao David
Roche Pharma Research and Early Development (pRED), Roche Innovation Center Basel, F. Hoffmann-La Roche Ltd., Grenzacherstrasse 124, CH-4070 Basel, Switzerland.
Institute of Bioengineering, School of Life Sciences, Ecole Polytechnique Fédérale de Lausanne (EPFL), Lausanne, 1015, Switzerland.
Comput Struct Biotechnol J. 2024 Jul 6;23:2872-2882. doi: 10.1016/j.csbj.2024.06.029. eCollection 2024 Dec.
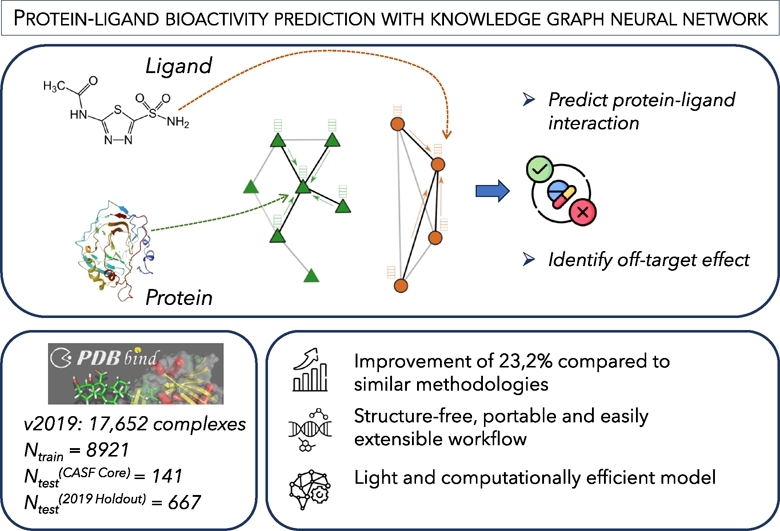
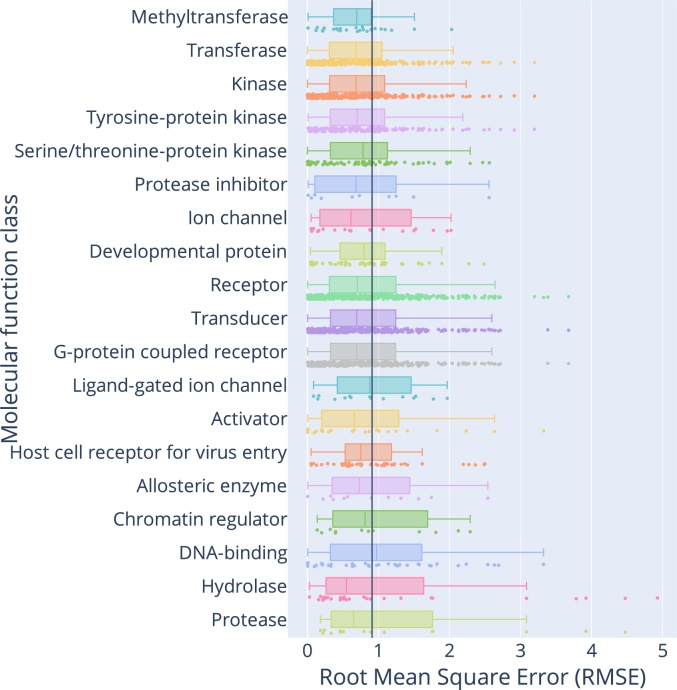
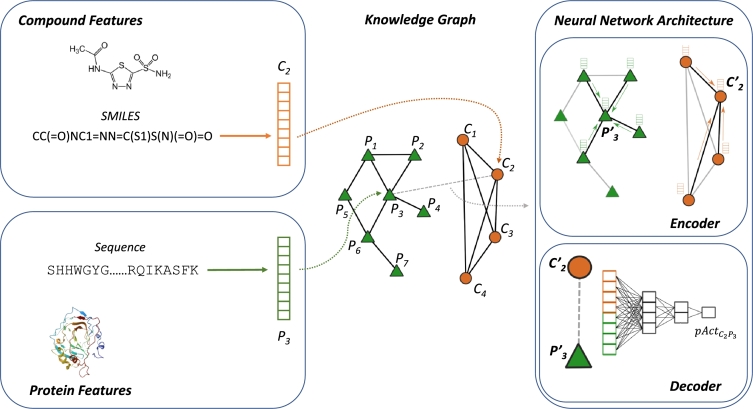
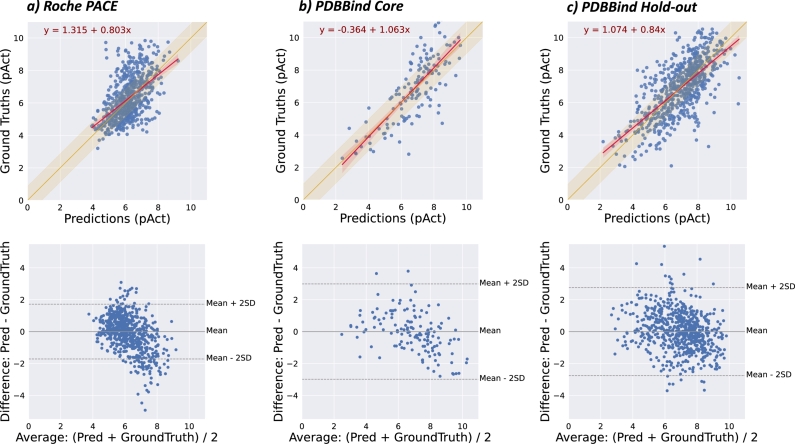
Protein-ligand interactions (PLIs) determine the efficacy and safety profiles of small molecule drugs. Existing methods rely on either structural information or resource-intensive computations to predict PLI, casting doubt on whether it is possible to perform structure-free PLI predictions at low computational cost. Here we show that a light-weight graph neural network (GNN), trained with quantitative PLIs of a small number of proteins and ligands, is able to predict the strength of unseen PLIs. The model has no direct access to structural information about the protein-ligand complexes. Instead, the predictive power is provided by encoding the entire chemical and proteomic space in a single heterogeneous graph, encapsulating primary protein sequence, gene expression, the protein-protein interaction network, and structural similarities between ligands. This novel approach performs competitively with, or better than, structure-aware models. Our results suggest that existing PLI prediction methods may be improved by incorporating representation learning techniques that embed biological and chemical knowledge.
蛋白质-配体相互作用(PLIs)决定了小分子药物的疗效和安全性。现有方法依靠结构信息或资源密集型计算来预测PLI,这让人怀疑是否有可能以低计算成本进行无结构的PLI预测。在这里,我们表明,一个轻量级图神经网络(GNN),通过少量蛋白质和配体的定量PLIs进行训练,能够预测未见过的PLIs的强度。该模型无法直接获取有关蛋白质-配体复合物的结构信息。相反,预测能力是通过在单个异构图中对整个化学和蛋白质组学空间进行编码来提供的,该图封装了主要蛋白质序列、基因表达、蛋白质-蛋白质相互作用网络以及配体之间的结构相似性。这种新方法的表现与结构感知模型相当,甚至更好。我们的结果表明,现有的PLI预测方法可能通过纳入嵌入生物和化学知识的表示学习技术来改进。