Diagnostic and Research Institute of Hygiene, Microbiology and Environmental Medicine, Medical University of Graz, Graz, Austria.
Institute for Infectious Diseases and Infection Control, Jena University Hospital, Jena, Germany.
J Clin Microbiol. 2024 Sep 11;62(9):e0062824. doi: 10.1128/jcm.00628-24. Epub 2024 Aug 19.
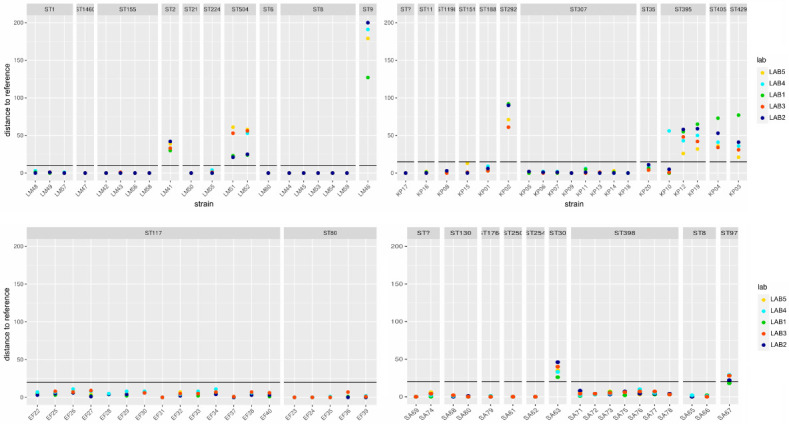
Nanopore sequencing has shown the potential to democratize genomic pathogen surveillance due to its ease of use and low entry cost. However, recent genotyping studies showed discrepant results compared to gold-standard short-read sequencing. Furthermore, although essential for widespread application, the reproducibility of nanopore-only genotyping remains largely unresolved. In our multicenter performance study involving five laboratories, four public health-relevant bacterial species were sequenced with the latest R10.4.1 flow cells and V14 chemistry. Core genome MLST analysis of over 500 data sets revealed highly strain-specific typing errors in all species in each laboratory. Investigation of the methylation-related errors revealed consistent DNA motifs at error-prone sites across participants at read level. Depending on the frequency of incorrect target reads, this either leads to correct or incorrect typing, whereby only minimal frequency deviations can randomly determine the final result. PCR preamplification, recent basecalling model updates and an optimized polishing strategy notably diminished the non-reproducible typing. Our study highlights the potential for new errors to appear with each newly sequenced strain and lays the foundation for computational approaches to reduce such typing errors. In conclusion, our multicenter study shows the necessity for a new validation concept for nanopore sequencing-based, standardized bacterial typing, where single nucleotide accuracy is critical.
纳米孔测序因其使用方便和入门成本低而显示出普及基因组病原体监测的潜力。然而,最近的基因分型研究结果与金标准短读测序结果不一致。此外,尽管对于广泛应用至关重要,但纳米孔单一组分基因分型的可重复性在很大程度上仍未得到解决。在我们涉及五个实验室的多中心性能研究中,使用最新的 R10.4.1 流池和 V14 化学物质对四个与公共卫生相关的细菌物种进行了测序。对 500 多个数据集的核心基因组 MLST 分析表明,在每个实验室的所有物种中,都存在高度菌株特异性的分型错误。对与甲基化相关的错误的研究表明,在阅读水平上,所有参与者在易错位点都存在一致的 DNA 基序。根据错误目标读取的频率,这要么导致正确的或错误的分型,其中只有最小的频率偏差可以随机确定最终结果。PCR 预扩增、最近的碱基调用模型更新和优化的后处理策略显著减少了不可重复的分型。我们的研究强调了每个新测序菌株都有可能出现新错误,并为减少这种分型错误的计算方法奠定了基础。总之,我们的多中心研究表明,有必要为基于纳米孔测序的标准化细菌分型建立新的验证概念,其中单核苷酸准确性至关重要。