National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD, 20894, USA.
Biochemistry and Biophysics Center, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD, 20892, USA.
Nat Commun. 2024 Aug 24;15(1):7296. doi: 10.1038/s41467-024-51801-z.
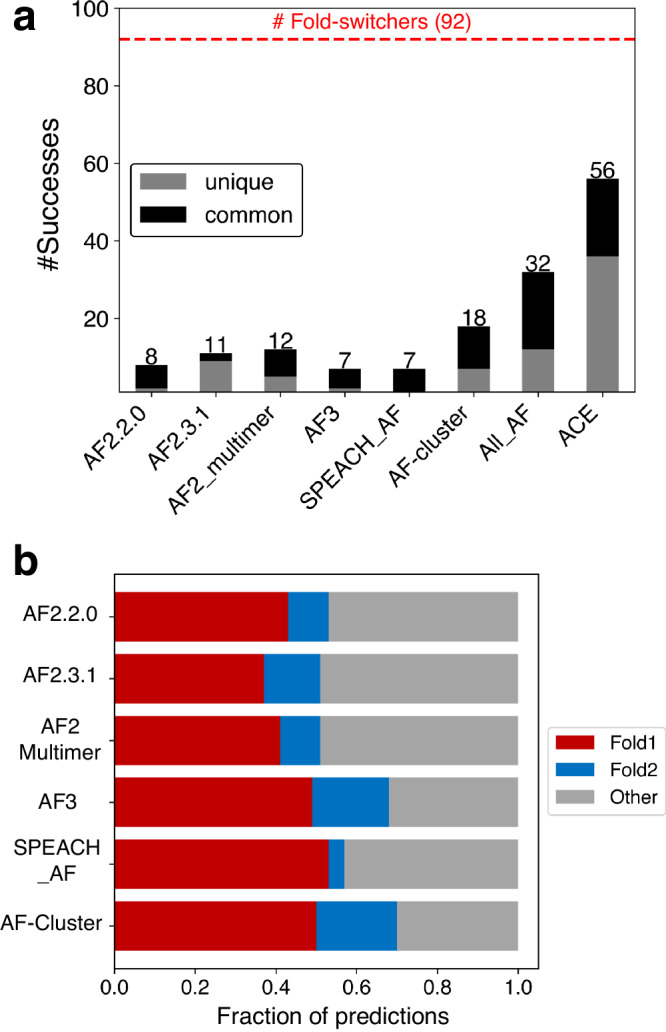
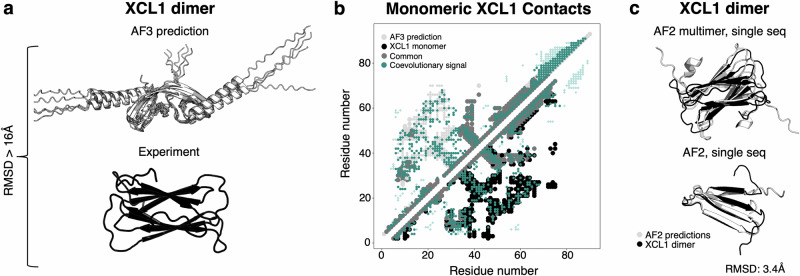
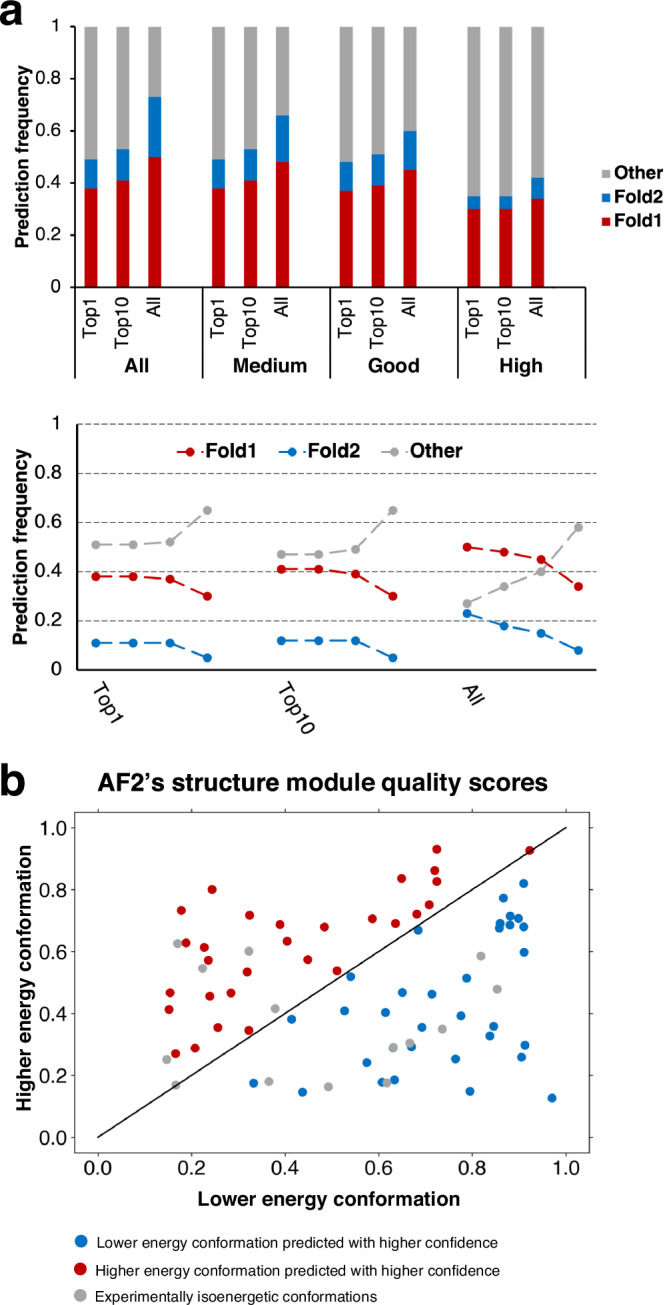
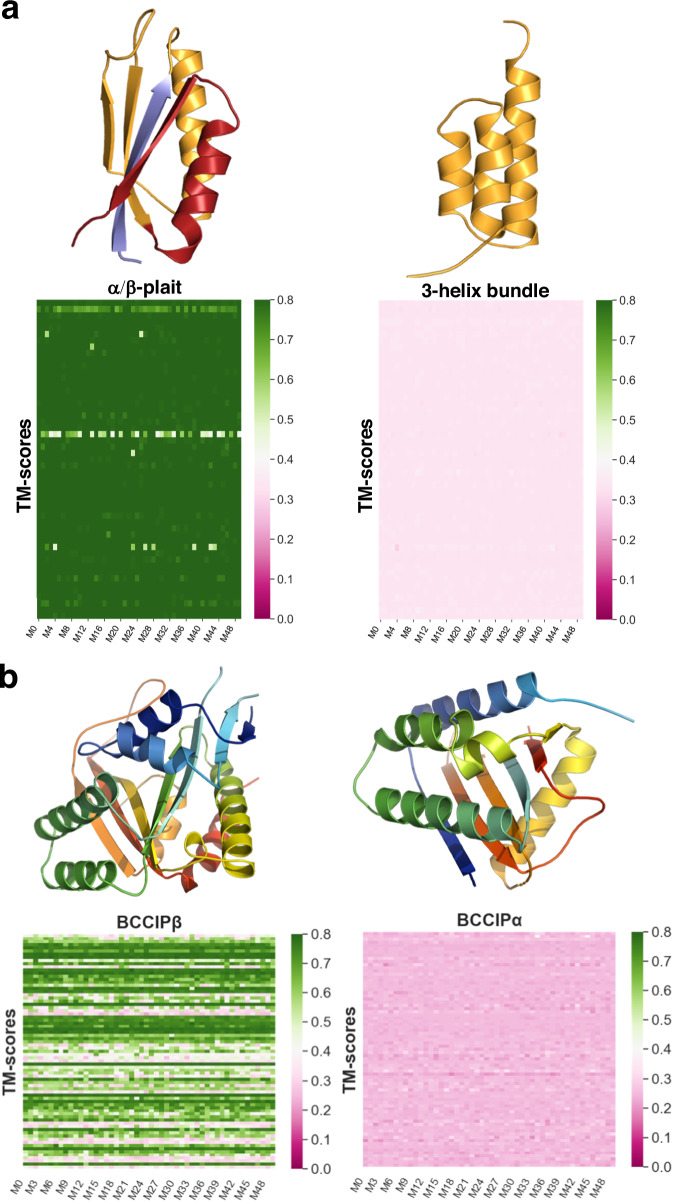
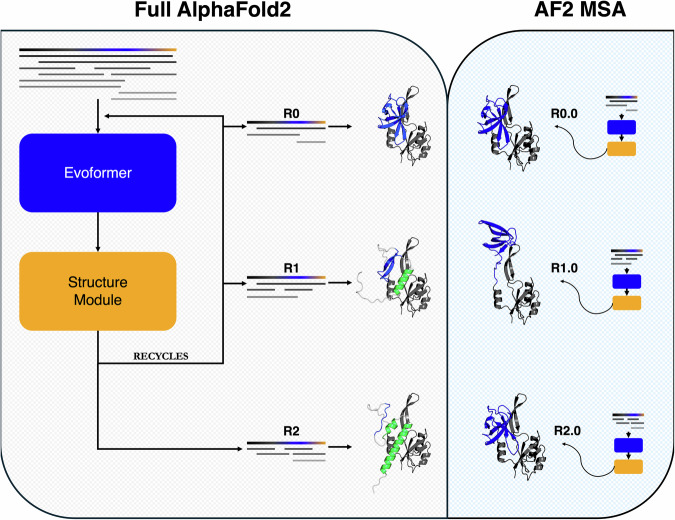
Recent work suggests that AlphaFold (AF)-a deep learning-based model that can accurately infer protein structure from sequence-may discern important features of folded protein energy landscapes, defined by the diversity and frequency of different conformations in the folded state. Here, we test the limits of its predictive power on fold-switching proteins, which assume two structures with regions of distinct secondary and/or tertiary structure. We find that (1) AF is a weak predictor of fold switching and (2) some of its successes result from memorization of training-set structures rather than learned protein energetics. Combining >280,000 models from several implementations of AF2 and AF3, a 35% success rate was achieved for fold switchers likely in AF's training sets. AF2's confidence metrics selected against models consistent with experimentally determined fold-switching structures and failed to discriminate between low and high energy conformations. Further, AF captured only one out of seven experimentally confirmed fold switchers outside of its training sets despite extensive sampling of an additional ~280,000 models. Several observations indicate that AF2 has memorized structural information during training, and AF3 misassigns coevolutionary restraints. These limitations constrain the scope of successful predictions, highlighting the need for physically based methods that readily predict multiple protein conformations.
最近的研究表明,基于深度学习的 AlphaFold(AF)模型可以从序列中准确推断蛋白质结构,它可能能够识别折叠蛋白质能量景观的重要特征,这些特征由折叠状态下不同构象的多样性和频率定义。在这里,我们测试了它在折叠转换蛋白上的预测能力的极限,折叠转换蛋白假设具有两个具有明显二级和/或三级结构的区域的结构。我们发现:(1)AF 是折叠转换的弱预测因子;(2)其某些成功源自于对训练集结构的记忆,而不是从学习到的蛋白质能量学中得到的。将来自几个 AF2 和 AF3 实现的超过 280,000 个模型组合起来,对于可能在 AF 训练集中的折叠转换蛋白,成功率达到了 35%。AF2 的置信度指标针对与实验确定的折叠转换结构一致的模型进行选择,并且无法区分低能和高能构象。此外,尽管对另外约 280,000 个模型进行了广泛的采样,AF 仅捕获了其训练集之外的七个实验确认的折叠转换蛋白中的一个。有几个观察结果表明,AF2 在训练过程中已经记住了结构信息,而 AF3 错误地分配了共进化限制。这些局限性限制了成功预测的范围,突出了需要易于预测多种蛋白质构象的基于物理的方法。